Identify samples with suspiciously high correlations and phenotype similarities
Usage
doppelgangR(
esets,
separator = ":",
corFinder.args = list(separator = separator, use.ComBat = TRUE, method = "pearson"),
phenoFinder.args = list(separator = separator, vectorDistFun = vectorWeightedDist),
outlierFinder.expr.args = list(bonf.prob = 0.5, transFun = atanh, tail = "upper"),
outlierFinder.pheno.args = list(normal.upper.thresh = 0.99, bonf.prob = NULL, tail =
"upper"),
smokingGunFinder.args = list(transFun = I),
impute.knn.args = list(k = 10, rowmax = 0.5, colmax = 0.8, maxp = 1500, rng.seed =
362436069),
manual.smokingguns = NULL,
automatic.smokingguns = FALSE,
within.datasets.only = FALSE,
intermediate.pruning = FALSE,
cache.dir = "cache",
BPPARAM = bpparam(),
verbose = TRUE
)Arguments
- esets
a list of ExpressionSets, containing the numeric and phenotypic data to be analyzed.
- separator
a delimitor to use between dataset names and sample names
- corFinder.args
a list of arguments to be passed to the corFinder function.
- phenoFinder.args
a list of arguments to be passed to the phenoFinder function. If NULL, samples with similar phenotypes will not be searched for.
- outlierFinder.expr.args
a list of arguments to be passed to outlierFinder when called for expression data
- outlierFinder.pheno.args
a list of arguments to be passed to outlierFinder when called for phenotype data
- smokingGunFinder.args
a list of arguments to be passed to smokingGunFinder
- impute.knn.args
a list of arguments to be passed to impute::impute.knn. Set to NULL to do no knn imputation.
- manual.smokingguns
a character vector of phenoData columns that, if identical, will be considered evidence of duplication
- automatic.smokingguns
automatically look for "smoking guns." If TRUE, look for phenotype variables that are unique to each patient in dataset 1, also unique to each patient in dataset 2, but contain exact matches between datasets 1 and 2.
- within.datasets.only
If TRUE, only search within each dataset for doppelgangers.
- intermediate.pruning
The default setting FALSE will result in output with no missing values, but uses extra memory because all results from the expression, phenotype, and smoking gun doppelganger searches must be saved until the end. Setting this to TRUE will save memory for very large searches, but distance metrics will only be available if that value was identified as a doppelganger (for example, phenotype doppelgangers will have missing values for the expression and smoking gun similarity).
- cache.dir
The name of a directory in which to cache or look up results to save re-calculating correlations. Set to NULL for no caching.
- BPPARAM
Argument for BiocParallel::bplapply(), by default will use all cores of a multi-core machine
- verbose
Print progress information
Examples
example("phenoFinder")
#>
#> phnFnd> library(curatedOvarianData)
#>
#> phnFnd> data(GSE32063_eset)
#>
#> phnFnd> data(GSE17260_eset)
#>
#> phnFnd> esets2 <- list(JapaneseB=GSE32063_eset,
#> phnFnd+ Yoshihara2010=GSE17260_eset)
#>
#> phnFnd> ## standardize the sample ids to improve matching based on clinical annotation
#> phnFnd> esets2 <- lapply(esets2, function(X){
#> phnFnd+ X$alt_sample_name <- paste(X$sample_type, gsub("[^0-9]", "", X$alt_sample_name), sep="_")
#> phnFnd+
#> phnFnd+ ## Removal of columns that cannot possibly match also helps duplicated patients to stand out
#> phnFnd+ pData(X) <- pData(X)[, !grepl("uncurated_author_metadata", colnames(pData(X)))]
#> phnFnd+ X <- X[, 1:20] ##speed computations
#> phnFnd+ return(X) })
#>
#> phnFnd> ## See first six samples in both rows and columns
#> phnFnd> phenoFinder(esets2)[1:6, 1:6]
#> GSM432220 GSM432221 GSM432222 GSM432223 GSM432224 GSM432225
#> GSM795125 0.2351904 0.1014047 0.3525417 0.7274151 0.2189890 0.27397077
#> GSM795126 0.5404524 0.2588727 0.4083015 0.4079720 0.2927870 0.74123368
#> GSM795127 0.3791279 0.5008562 0.4983502 0.4981226 0.6385506 0.04416984
#> GSM795128 0.2351904 0.1014047 0.3525417 0.3523760 0.2189890 0.27397077
#> GSM795129 0.1076309 0.2395470 0.2190910 0.2189890 0.3643260 0.16030839
#> GSM795130 0.2603947 0.1344290 0.1077761 0.1076793 0.2489234 0.29544860
results2 <- doppelgangR(esets2, cache.dir = NULL)
#> Finalizing...
results2
#> S4 object of class: DoppelGang
#> Number of potential doppelgangers: 6 : 6 expression, 2 phenotype, 0 smoking gun.
#>
#> Use summary(object) to obtain a data.frame of potential doppelgangrs.
#>
#>
plot(results2)
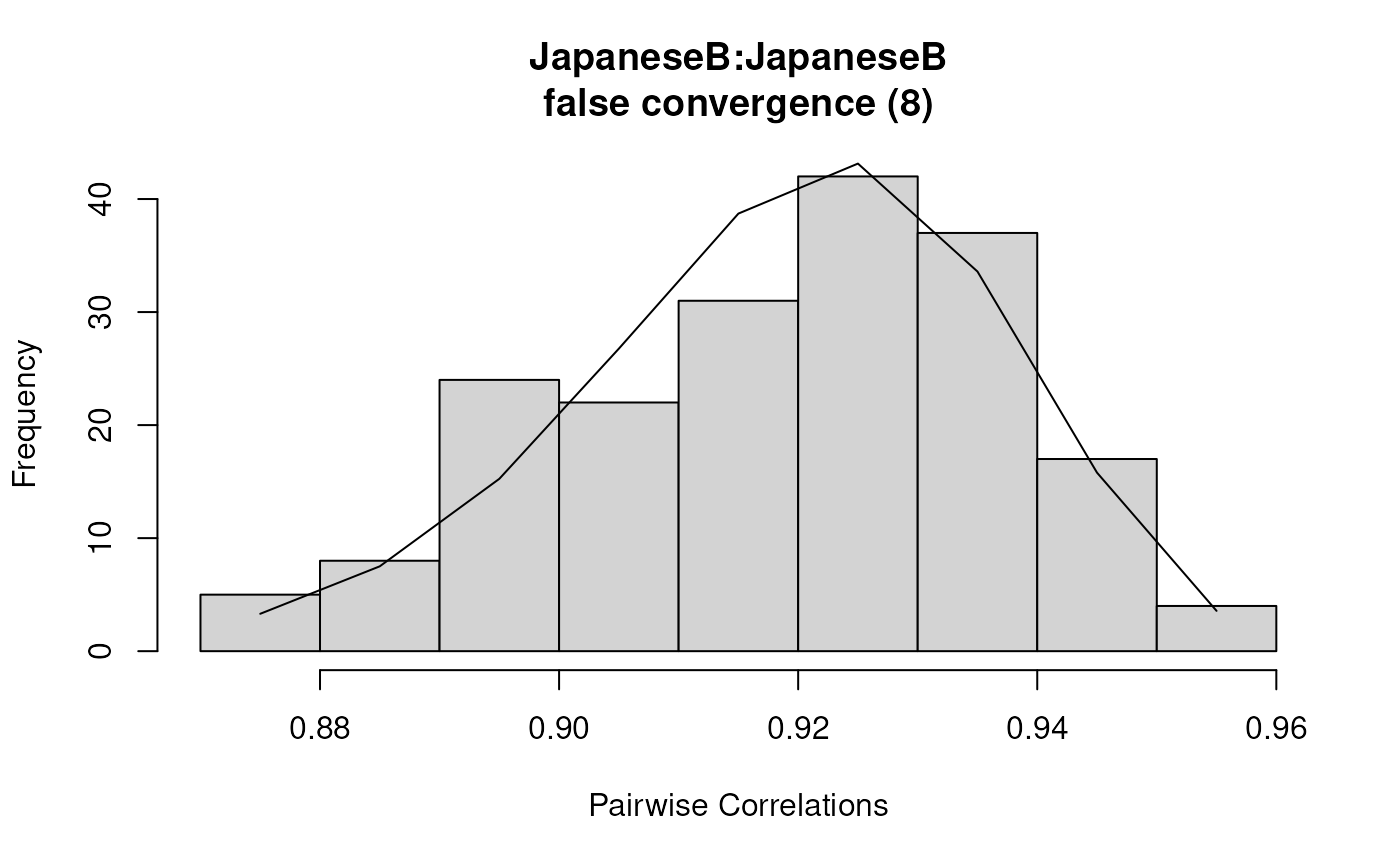
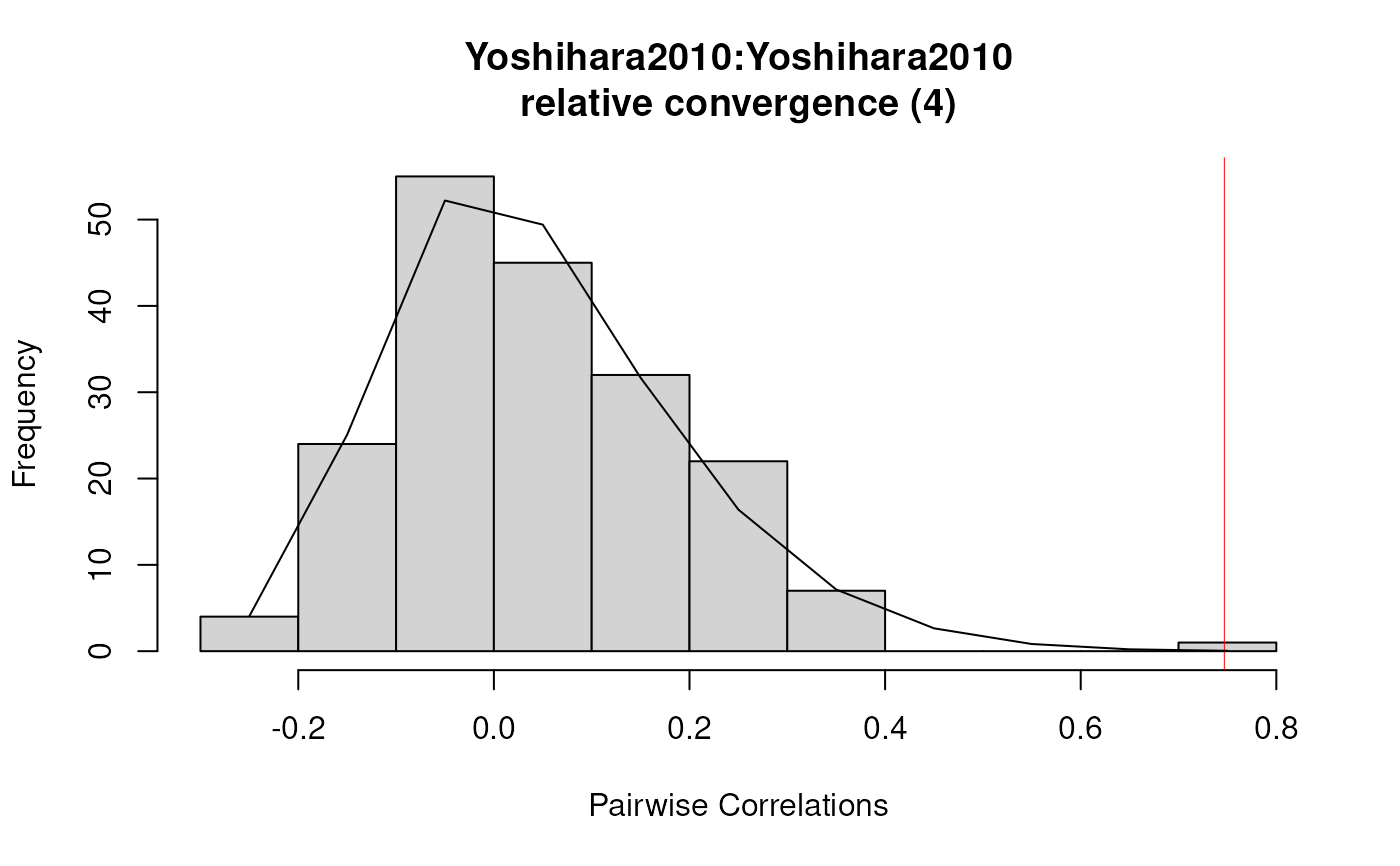
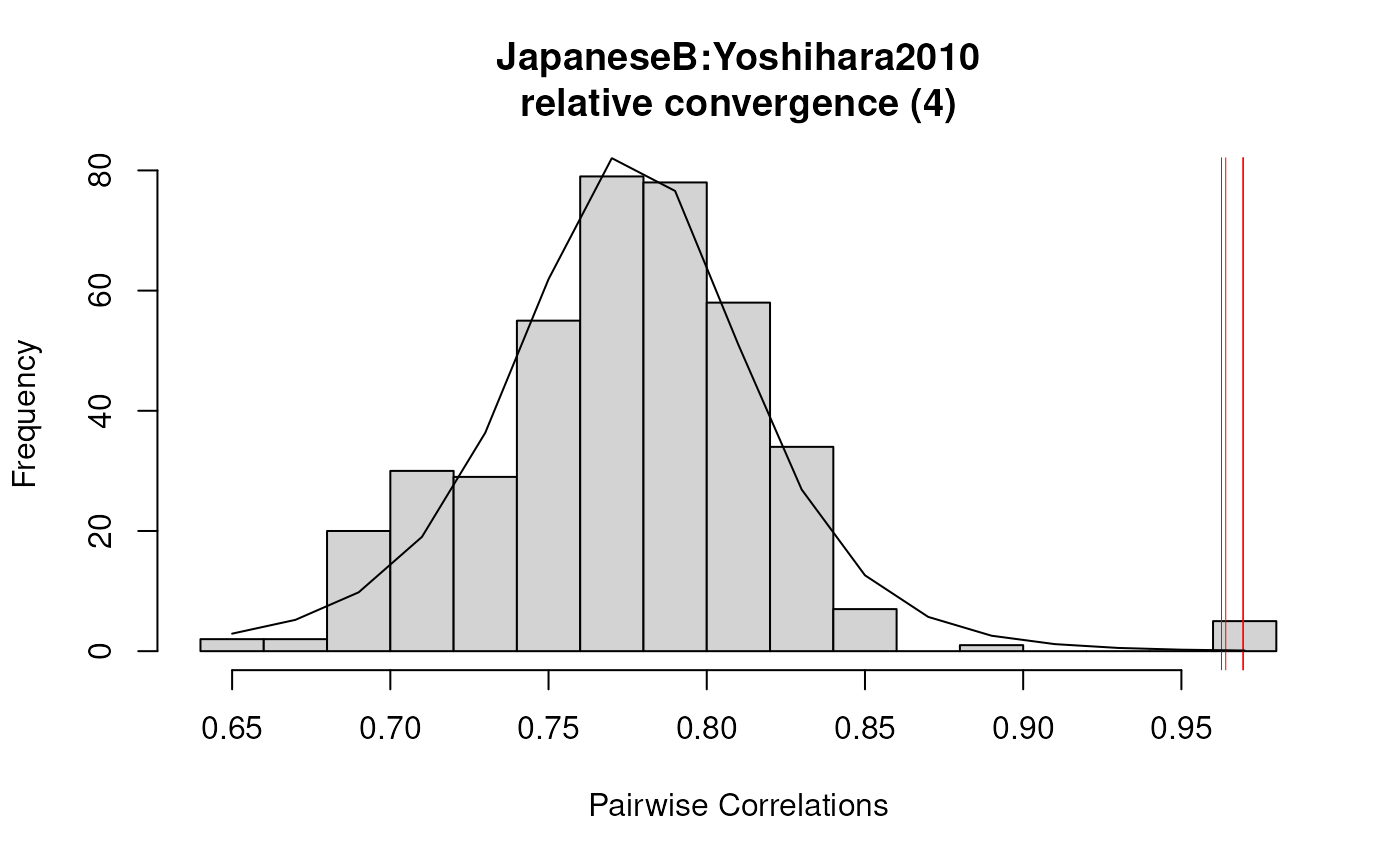 summary(results2)
#> sample1 sample2 expr.similarity expr.doppel
#> 1 Yoshihara2010:GSM432230 Yoshihara2010:GSM432231 0.7468418 TRUE
#> 2 JapaneseB:GSM795125 Yoshihara2010:GSM432223 0.9694795 TRUE
#> 3 JapaneseB:GSM795126 Yoshihara2010:GSM432225 0.9626765 TRUE
#> 4 JapaneseB:GSM795127 Yoshihara2010:GSM432226 0.9695461 TRUE
#> 5 JapaneseB:GSM795128 Yoshihara2010:GSM432228 0.9640154 TRUE
#> 6 JapaneseB:GSM795129 Yoshihara2010:GSM432229 0.9695206 TRUE
#> pheno.similarity pheno.doppel smokinggun.similarity smokinggun.doppel
#> 1 0.2029299 FALSE NA FALSE
#> 2 0.7274151 FALSE NA FALSE
#> 3 0.7412337 FALSE NA FALSE
#> 4 1.0000000 TRUE NA FALSE
#> 5 0.5398956 FALSE NA FALSE
#> 6 1.0000000 TRUE NA FALSE
#> alt_sample_name unique_patient_ID sample_type histological_type
#> 1 tumor_112:tumor_113 NA:NA tumor:tumor ser:ser
#> 2 tumor_106:tumor_106 NA:NA tumor:tumor ser:ser
#> 3 tumor_108:tumor_108 NA:NA tumor:tumor ser:ser
#> 4 tumor_109:tumor_109 NA:NA tumor:tumor ser:ser
#> 5 tumor_110:tumor_110 NA:NA tumor:tumor ser:ser
#> 6 tumor_111:tumor_111 NA:NA tumor:tumor ser:ser
#> primarysite arrayedsite summarygrade summarystage tumorstage substage grade
#> 1 ov:ov NA:NA low:low late:late 3:3 a:c 2:1
#> 2 NA:ov NA:NA low:high late:late 3:3 c:c 2:3
#> 3 NA:ov NA:NA high:low late:late 3:3 b:b 3:1
#> 4 NA:ov NA:NA high:high late:late 3:3 c:c 3:3
#> 5 NA:ov NA:NA low:high late:late 3:3 c:c 2:3
#> 6 NA:ov NA:NA low:low late:late 3:3 c:c 2:2
#> age_at_initial_pathologic_diagnosis pltx tax neo days_to_tumor_recurrence
#> 1 NA:NA y:y y:y NA:NA 1440:1290
#> 2 NA:NA y:y y:y NA:NA NA:450
#> 3 NA:NA y:y y:y NA:NA NA:1110
#> 4 NA:NA y:y y:y NA:NA NA:210
#> 5 NA:NA y:y y:y NA:NA NA:1800
#> 6 NA:NA y:y y:y NA:NA NA:540
#> recurrence_status days_to_death vital_status os_binary
#> 1 norecurrence:recurrence 1440:2070 living:living NA:NA
#> 2 NA:recurrence 780:780 living:living NA:NA
#> 3 NA:norecurrence 1110:1110 living:living NA:NA
#> 4 NA:recurrence 600:600 deceased:deceased NA:NA
#> 5 NA:norecurrence 2910:1800 living:living NA:NA
#> 6 NA:recurrence 1710:1710 deceased:deceased NA:NA
#> relapse_binary site_of_tumor_first_recurrence primary_therapy_outcome_success
#> 1 NA:NA NA:NA NA:NA
#> 2 NA:NA NA:NA NA:NA
#> 3 NA:NA NA:NA NA:NA
#> 4 NA:NA NA:NA NA:NA
#> 5 NA:NA NA:NA NA:NA
#> 6 NA:NA NA:NA NA:NA
#> debulking percent_normal_cells percent_stromal_cells
#> 1 optimal:suboptimal NA:NA NA:NA
#> 2 suboptimal:suboptimal NA:NA NA:NA
#> 3 optimal:optimal NA:NA NA:NA
#> 4 suboptimal:suboptimal NA:NA NA:NA
#> 5 suboptimal:suboptimal NA:NA NA:NA
#> 6 suboptimal:suboptimal NA:NA NA:NA
#> percent_tumor_cells batch flag flag_notes
#> 1 NA:NA NA:NA NA:NA NA:NA
#> 2 NA:NA NA:NA NA:NA NA:NA
#> 3 NA:NA NA:NA NA:NA NA:NA
#> 4 NA:NA NA:NA NA:NA NA:NA
#> 5 NA:NA NA:NA NA:NA NA:NA
#> 6 NA:NA NA:NA NA:NA NA:NA
## Set phenoFinder.args=NULL to ignore similar phenotypes, and
## turn off ComBat batch correction:
if (FALSE) { # \dontrun{
results2 <- doppelgangR(testesets,
corFinder.args=list(use.ComBat=FALSE), phenoFinder.args=NULL,
cache.dir=NULL)
summary(results2)
library(curatedOvarianData)
data(GSE32062.GPL6480_eset)
data(GSE32063_eset)
data(GSE12470_eset)
data(GSE17260_eset)
testesets <- list(JapaneseA = GSE32062.GPL6480_eset,
JapaneseB = GSE32063_eset,
Yoshihara2009 = GSE12470_eset,
Yoshihara2010 = GSE17260_eset)
## standardize the sample ids to improve matching
## based on clinical annotation
testesets <- lapply(testesets, function(X) {
X$alt_sample_name <-
paste(X$sample_type, gsub("[^0-9]", "", X$alt_sample_name), sep = "_")
pData(X) <-
pData(X)[,!grepl("uncurated_author_metadata", colnames(pData(X)))]
X[, 1:20] ##speed computations
})
(results1 <- doppelgangR(testesets, cache.dir = NULL))
plot(results1)
summary(results1)
} # }
summary(results2)
#> sample1 sample2 expr.similarity expr.doppel
#> 1 Yoshihara2010:GSM432230 Yoshihara2010:GSM432231 0.7468418 TRUE
#> 2 JapaneseB:GSM795125 Yoshihara2010:GSM432223 0.9694795 TRUE
#> 3 JapaneseB:GSM795126 Yoshihara2010:GSM432225 0.9626765 TRUE
#> 4 JapaneseB:GSM795127 Yoshihara2010:GSM432226 0.9695461 TRUE
#> 5 JapaneseB:GSM795128 Yoshihara2010:GSM432228 0.9640154 TRUE
#> 6 JapaneseB:GSM795129 Yoshihara2010:GSM432229 0.9695206 TRUE
#> pheno.similarity pheno.doppel smokinggun.similarity smokinggun.doppel
#> 1 0.2029299 FALSE NA FALSE
#> 2 0.7274151 FALSE NA FALSE
#> 3 0.7412337 FALSE NA FALSE
#> 4 1.0000000 TRUE NA FALSE
#> 5 0.5398956 FALSE NA FALSE
#> 6 1.0000000 TRUE NA FALSE
#> alt_sample_name unique_patient_ID sample_type histological_type
#> 1 tumor_112:tumor_113 NA:NA tumor:tumor ser:ser
#> 2 tumor_106:tumor_106 NA:NA tumor:tumor ser:ser
#> 3 tumor_108:tumor_108 NA:NA tumor:tumor ser:ser
#> 4 tumor_109:tumor_109 NA:NA tumor:tumor ser:ser
#> 5 tumor_110:tumor_110 NA:NA tumor:tumor ser:ser
#> 6 tumor_111:tumor_111 NA:NA tumor:tumor ser:ser
#> primarysite arrayedsite summarygrade summarystage tumorstage substage grade
#> 1 ov:ov NA:NA low:low late:late 3:3 a:c 2:1
#> 2 NA:ov NA:NA low:high late:late 3:3 c:c 2:3
#> 3 NA:ov NA:NA high:low late:late 3:3 b:b 3:1
#> 4 NA:ov NA:NA high:high late:late 3:3 c:c 3:3
#> 5 NA:ov NA:NA low:high late:late 3:3 c:c 2:3
#> 6 NA:ov NA:NA low:low late:late 3:3 c:c 2:2
#> age_at_initial_pathologic_diagnosis pltx tax neo days_to_tumor_recurrence
#> 1 NA:NA y:y y:y NA:NA 1440:1290
#> 2 NA:NA y:y y:y NA:NA NA:450
#> 3 NA:NA y:y y:y NA:NA NA:1110
#> 4 NA:NA y:y y:y NA:NA NA:210
#> 5 NA:NA y:y y:y NA:NA NA:1800
#> 6 NA:NA y:y y:y NA:NA NA:540
#> recurrence_status days_to_death vital_status os_binary
#> 1 norecurrence:recurrence 1440:2070 living:living NA:NA
#> 2 NA:recurrence 780:780 living:living NA:NA
#> 3 NA:norecurrence 1110:1110 living:living NA:NA
#> 4 NA:recurrence 600:600 deceased:deceased NA:NA
#> 5 NA:norecurrence 2910:1800 living:living NA:NA
#> 6 NA:recurrence 1710:1710 deceased:deceased NA:NA
#> relapse_binary site_of_tumor_first_recurrence primary_therapy_outcome_success
#> 1 NA:NA NA:NA NA:NA
#> 2 NA:NA NA:NA NA:NA
#> 3 NA:NA NA:NA NA:NA
#> 4 NA:NA NA:NA NA:NA
#> 5 NA:NA NA:NA NA:NA
#> 6 NA:NA NA:NA NA:NA
#> debulking percent_normal_cells percent_stromal_cells
#> 1 optimal:suboptimal NA:NA NA:NA
#> 2 suboptimal:suboptimal NA:NA NA:NA
#> 3 optimal:optimal NA:NA NA:NA
#> 4 suboptimal:suboptimal NA:NA NA:NA
#> 5 suboptimal:suboptimal NA:NA NA:NA
#> 6 suboptimal:suboptimal NA:NA NA:NA
#> percent_tumor_cells batch flag flag_notes
#> 1 NA:NA NA:NA NA:NA NA:NA
#> 2 NA:NA NA:NA NA:NA NA:NA
#> 3 NA:NA NA:NA NA:NA NA:NA
#> 4 NA:NA NA:NA NA:NA NA:NA
#> 5 NA:NA NA:NA NA:NA NA:NA
#> 6 NA:NA NA:NA NA:NA NA:NA
## Set phenoFinder.args=NULL to ignore similar phenotypes, and
## turn off ComBat batch correction:
if (FALSE) { # \dontrun{
results2 <- doppelgangR(testesets,
corFinder.args=list(use.ComBat=FALSE), phenoFinder.args=NULL,
cache.dir=NULL)
summary(results2)
library(curatedOvarianData)
data(GSE32062.GPL6480_eset)
data(GSE32063_eset)
data(GSE12470_eset)
data(GSE17260_eset)
testesets <- list(JapaneseA = GSE32062.GPL6480_eset,
JapaneseB = GSE32063_eset,
Yoshihara2009 = GSE12470_eset,
Yoshihara2010 = GSE17260_eset)
## standardize the sample ids to improve matching
## based on clinical annotation
testesets <- lapply(testesets, function(X) {
X$alt_sample_name <-
paste(X$sample_type, gsub("[^0-9]", "", X$alt_sample_name), sep = "_")
pData(X) <-
pData(X)[,!grepl("uncurated_author_metadata", colnames(pData(X)))]
X[, 1:20] ##speed computations
})
(results1 <- doppelgangR(testesets, cache.dir = NULL))
plot(results1)
summary(results1)
} # }