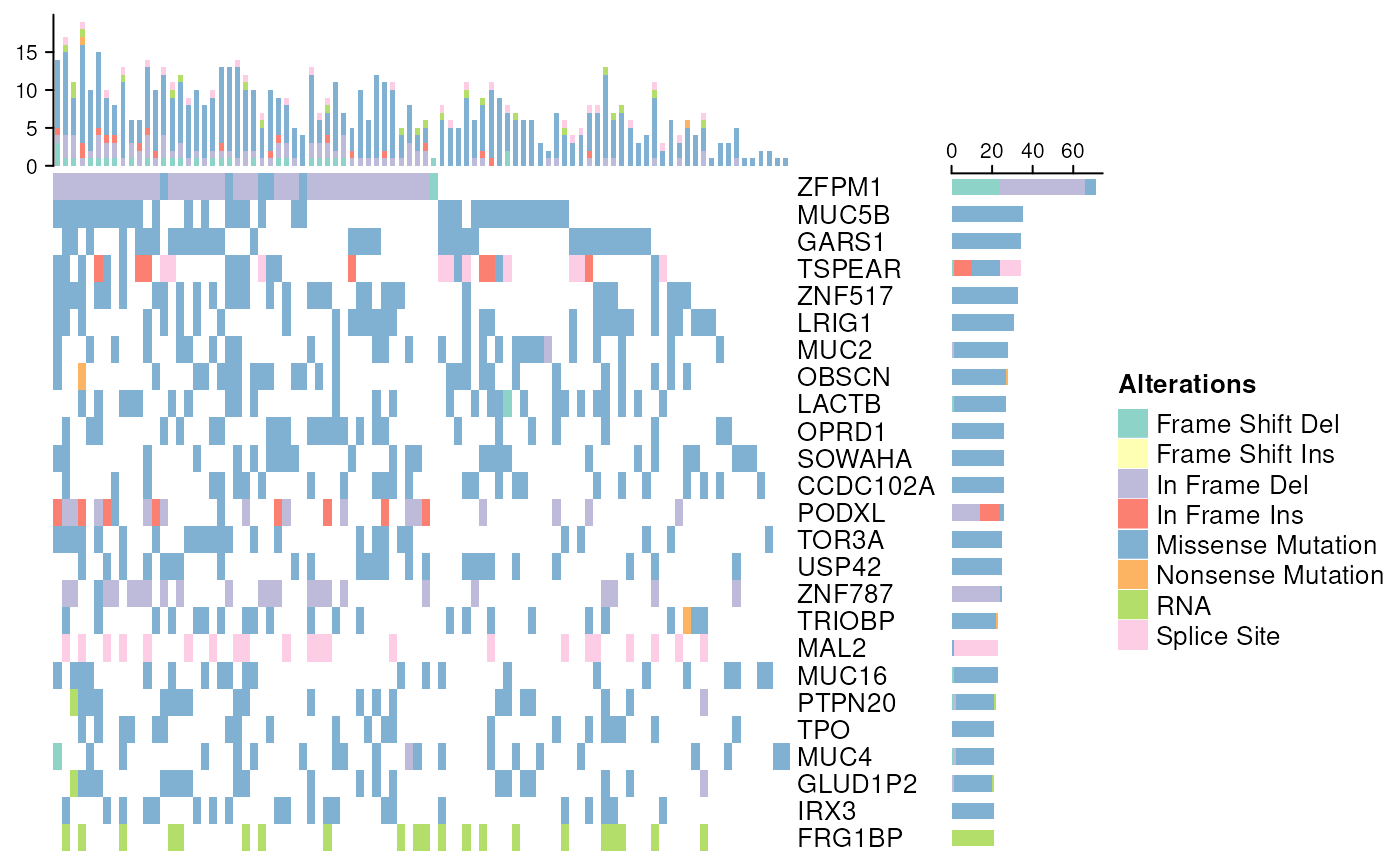
OncoPrint for TCGA Mutation Assays
Usage
oncoPrintTCGA(
multiassayexperiment,
matchassay = "*_Mutation-*",
variantCol = "Variant_Classification",
brewerPal = "Set3",
ntop = 25,
incl.thresh = 0.01,
rowcol = "Hugo_Symbol"
)Arguments
- multiassayexperiment
A
MultiAssayExperiment, usually fromcuratedTCGAData- matchassay
character(1) The name of the assay containing mutation data, this can be a pattern (e.g., "_Mutation-", the default)
- variantCol
character(1) The name of the metadata column containing the mutation categories, usually "Variant_Classification" in TCGA
- brewerPal
character(1) The name of the
RColorBrewer::brewer.palpalette, (default: "Set3")- ntop
integer(1) The number of the top N genes for displaying based on per-sample mutation frequency
- incl.thresh
double(1) The inclusion threshold for empirical mutations, mutations less frequent than this value will not be included
- rowcol
character(1) The name of the column in the metadata to annotate the rows with either "Hugo_Symbol" (default) or
Examples
library(curatedTCGAData)
acc <- curatedTCGAData("ACC", "Mutation", version = "1.1.38", FALSE)
#> Querying and downloading: ACC_Mutation-20160128
#> see ?curatedTCGAData and browseVignettes('curatedTCGAData') for documentation
#> loading from cache
#> Querying and downloading: ACC_colData-20160128
#> see ?curatedTCGAData and browseVignettes('curatedTCGAData') for documentation
#> loading from cache
#> Querying and downloading: ACC_metadata-20160128
#> see ?curatedTCGAData and browseVignettes('curatedTCGAData') for documentation
#> loading from cache
#> Querying and downloading: ACC_sampleMap-20160128
#> see ?curatedTCGAData and browseVignettes('curatedTCGAData') for documentation
#> loading from cache
#> harmonizing input:
#> removing 915 sampleMap rows not in names(experiments)
#> removing 2 colData rownames not in sampleMap 'primary'
oncoPrintTCGA(acc)
#> 24 genes were dropped because they have exons located on both strands of the
#> same reference sequence or on more than one reference sequence, so cannot be
#> represented by a single genomic range.
#> Use 'single.strand.genes.only=FALSE' to get all the genes in a GRangesList
#> object, or use suppressMessages() to suppress this message.
#> Warning: cannot switch some hg19's seqlevels from UCSC to NCBI style
#>
#> 'select()' returned 1:1 mapping between keys and columns
#> All mutation types: Frame Shift Del, Frame Shift Ins, In Frame Del, In
#> Frame Ins, Missense Mutation, Nonsense Mutation, RNA, Splice Site.
#> `alter_fun` is assumed vectorizable. If it does not generate correct
#> plot, please set `alter_fun_is_vectorized = FALSE` in `oncoPrint()`.