TCGAutils: Helper functions for working with TCGA datasets
Marcel Ramos & Levi Waldron
July 02, 2026
Source:vignettes/TCGAutils.Rmd
TCGAutils.RmdOverview
The TCGAutils package completes a suite of Bioconductor
packages for convenient access, integration, and analysis of The
Cancer Genome Atlas. It includes: 0. helpers for working with TCGA
through the Bioconductor packages MultiAssayExperiment
(for coordinated representation and manipulation of multi-omits
experiments) and curatedTCGAData,
which provides unrestricted TCGA data as
MultiAssayExperiment objects, 0. helpers for importing TCGA
data as from flat data structures such as data.frame or
DataFrame read from delimited data structures provided by
the Broad Institute’s Firehose, GenomicDataCommons, and 0. functions for
interpreting TCGA barcodes and for mapping between barcodes and
Universally Unique Identifiers (UUIDs).
Installation
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("TCGAutils")Required packages for this vignette:
library(TCGAutils)
library(curatedTCGAData)
library(MultiAssayExperiment)
library(RTCGAToolbox)
library(rtracklayer)
library(R.utils)
library(GenomeInfoDb)
curatedTCGAData utility functions
Functions such as getSubtypeMap and
getClinicalNames provide information on data inside a
MultiAssayExperiment
object downloaded from curatedTCGAData.
sampleTables and TCGAsplitAssays support
useful operations on these MultiAssayExperiment
objects.
obtaining TCGA as MultiAssayExperiment objects from
curatedTCGAData
For a list of all available data types, use
dry.run = TRUE and an asterisk * as the assay
input value:
curatedTCGAData("COAD", "*", dry.run = TRUE, version = "1.1.38")## See '?curatedTCGAData' for 'diseaseCode' and 'assays' inputs## Querying EH with: COAD_CNASeq-20160128## Querying EH with: COAD_CNASNP-20160128## Querying EH with: COAD_CNVSNP-20160128## Querying EH with: COAD_GISTIC_AllByGene-20160128## Querying EH with: COAD_GISTIC_Peaks-20160128## Querying EH with: COAD_GISTIC_ThresholdedByGene-20160128## Querying EH with: COAD_Methylation_methyl27-20160128_assays## Querying EH with: COAD_Methylation_methyl27-20160128_se## Querying EH with: COAD_Methylation_methyl450-20160128_assays## Querying EH with: COAD_Methylation_methyl450-20160128_se## Querying EH with: COAD_miRNASeqGene-20160128## Querying EH with: COAD_mRNAArray-20160128## Querying EH with: COAD_Mutation-20160128## Querying EH with: COAD_RNASeq2GeneNorm-20160128## Querying EH with: COAD_RNASeqGene-20160128## Querying EH with: COAD_RPPAArray-20160128## ah_id title file_size
## 1 EH625 COAD_CNASeq-20160128 0.3 Mb
## 2 EH626 COAD_CNASNP-20160128 3.9 Mb
## 3 EH627 COAD_CNVSNP-20160128 0.9 Mb
## 4 EH629 COAD_GISTIC_AllByGene-20160128 0.5 Mb
## 5 EH2132 COAD_GISTIC_Peaks-20160128 0 Mb
## 6 EH630 COAD_GISTIC_ThresholdedByGene-20160128 0.3 Mb
## 7 EH2133 COAD_Methylation_methyl27-20160128_assays 37.2 Mb
## 8 EH2134 COAD_Methylation_methyl27-20160128_se 0.4 Mb
## 9 EH2135 COAD_Methylation_methyl450-20160128_assays 983.8 Mb
## 10 EH2136 COAD_Methylation_methyl450-20160128_se 6.1 Mb
## 11 EH634 COAD_miRNASeqGene-20160128 0.2 Mb
## 12 EH635 COAD_mRNAArray-20160128 8.1 Mb
## 13 EH636 COAD_Mutation-20160128 1.2 Mb
## 14 EH637 COAD_RNASeq2GeneNorm-20160128 8.8 Mb
## 15 EH638 COAD_RNASeqGene-20160128 0.4 Mb
## 16 EH639 COAD_RPPAArray-20160128 0.6 Mb
## rdataclass rdatadateadded rdatadateremoved
## 1 RaggedExperiment 2017-10-10 <NA>
## 2 RaggedExperiment 2017-10-10 <NA>
## 3 RaggedExperiment 2017-10-10 <NA>
## 4 SummarizedExperiment 2017-10-10 <NA>
## 5 RangedSummarizedExperiment 2019-01-09 <NA>
## 6 SummarizedExperiment 2017-10-10 <NA>
## 7 SummarizedExperiment 2019-01-09 <NA>
## 8 SummarizedExperiment 2019-01-09 <NA>
## 9 RaggedExperiment 2019-01-09 <NA>
## 10 SummarizedExperiment 2019-01-09 <NA>
## 11 SummarizedExperiment 2017-10-10 <NA>
## 12 SummarizedExperiment 2017-10-10 <NA>
## 13 RaggedExperiment 2017-10-10 <NA>
## 14 SummarizedExperiment 2017-10-10 <NA>
## 15 SummarizedExperiment 2017-10-10 <NA>
## 16 SummarizedExperiment 2017-10-10 <NA>In this example, we download part of the Colon Adenocarcinoma (COAD)
dataset using curatedTCGAData via
ExperimentHub. This command will download data types such
as CNASeq, Mutation, etc.:
coad <- curatedTCGAData(
diseaseCode = "COAD", assays = c("CNASeq", "Mutation", "miRNA*",
"RNASeq2*", "mRNAArray", "Methyl*"), version = "1.1.38", dry.run = FALSE
)
sampleTables: what sample types are present in the
data?
The sampleTables function gives a tally of available
samples in the dataset based on the TCGA barcode information.
sampleTables(coad)## $`COAD_CNASeq-20160128`
##
## 01 10 11
## 68 55 13
##
## $`COAD_miRNASeqGene-20160128`
##
## 01 02
## 220 1
##
## $`COAD_mRNAArray-20160128`
##
## 01 11
## 153 19
##
## $`COAD_Mutation-20160128`
##
## 01
## 154
##
## $`COAD_RNASeq2GeneNorm-20160128`
##
## 01
## 191
##
## $`COAD_Methylation_methyl27-20160128`
##
## 01 11
## 165 37
##
## $`COAD_Methylation_methyl450-20160128`
##
## 01 02 06 11
## 293 1 1 38For reference in interpreting the sample type codes, see the
sampleTypes table:
data("sampleTypes")
head(sampleTypes)## Code Definition Short.Letter.Code
## 1 01 Primary Solid Tumor TP
## 2 02 Recurrent Solid Tumor TR
## 3 03 Primary Blood Derived Cancer - Peripheral Blood TB
## 4 04 Recurrent Blood Derived Cancer - Bone Marrow TRBM
## 5 05 Additional - New Primary TAP
## 6 06 Metastatic TM
TCGAsplitAssays: separate the data from different
tissue types
TCGA datasets include multiple -omics for solid tumors, adjacent
normal tissues, blood-derived cancers and normals, and other tissue
types, which may be mixed together in a single dataset. The
MultiAssayExperiment object generated here has one patient
per row of its colData, but each patient may have two or
more -omics profiles by any assay, whether due to assaying of different
types of tissues or to technical replication.
TCGAsplitAssays separates profiles from different tissue
types (such as tumor and adjacent normal) into different assays of the
MultiAssayExperiment by taking a vector of sample codes,
and partitioning the current assays into assays with an appended sample
code:
(tnmae <- TCGAsplitAssays(coad, c("01", "11")))## Warning: Some 'sampleCodes' not found in assays## A MultiAssayExperiment object of 11 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 11:
## [1] 01_COAD_CNASeq-20160128: RaggedExperiment with 40530 rows and 68 columns
## [2] 11_COAD_CNASeq-20160128: RaggedExperiment with 40530 rows and 13 columns
## [3] 01_COAD_miRNASeqGene-20160128: SummarizedExperiment with 705 rows and 220 columns
## [4] 01_COAD_mRNAArray-20160128: SummarizedExperiment with 17814 rows and 153 columns
## [5] 11_COAD_mRNAArray-20160128: SummarizedExperiment with 17814 rows and 19 columns
## [6] 01_COAD_Mutation-20160128: RaggedExperiment with 62530 rows and 154 columns
## [7] 01_COAD_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 191 columns
## [8] 01_COAD_Methylation_methyl27-20160128: SummarizedExperiment with 27578 rows and 165 columns
## [9] 11_COAD_Methylation_methyl27-20160128: SummarizedExperiment with 27578 rows and 37 columns
## [10] 01_COAD_Methylation_methyl450-20160128: SummarizedExperiment with 485577 rows and 293 columns
## [11] 11_COAD_Methylation_methyl450-20160128: SummarizedExperiment with 485577 rows and 38 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesThe MultiAssayExperiment
package then provides functionality to merge replicate profiles for a
single patient (mergeReplicates()), which would now be
appropriate but would not have been appropriate before
splitting different tissue types into different assays, because that
would average measurements from tumors and normal tissues.
MultiAssayExperiment also defines the
MatchedAssayExperiment class, which eliminates any profiles
not present across all assays and ensures identical ordering of profiles
(columns) in each assay. In this example, it will match tumors to
adjacent normals in subsequent assays:
## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 853 sampleMap rows not in names(experiments)
## removing 260 colData rownames not in sampleMap 'primary'## A MatchedAssayExperiment object of 3 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 3:
## [1] 01_COAD_mRNAArray-20160128: SummarizedExperiment with 17814 rows and 138 columns
## [2] 01_COAD_Mutation-20160128: RaggedExperiment with 62530 rows and 138 columns
## [3] 01_COAD_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 138 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesOnly about 12 participants have both a matched tumor and solid normal sample.
getSubtypeMap: manually curated molecular subtypes
Per-tumor subtypes are saved in the metadata of the
colData slot of MultiAssayExperiment objects
downloaded from curatedTCGAData. These subtypes were
manually curated from the supplemental tables of all primary TCGA
publications:
getSubtypeMap(coad)## COAD_annotations COAD_subtype
## 1 Patient_ID patientID
## 2 msi MSI_status
## 3 methylation_subtypes methylation_subtype
## 4 mrna_subtypes expression_subtype
## 5 histological_subtypes histological_type
getClinicalNames: key “level 4” clinical &
pathological data
The curatedTCGAData colData contain
hundreds of columns, obtained from merging all unrestricted levels of
clinical, pathological, and biospecimen data. This function provides the
names of “level 4” clinical/pathological variables, which are the only
ones provided by most other TCGA analysis tools. Users may then use
these variable names for subsetting or analysis, and may even want to
subset the colData to only these commonly used
variables.
getClinicalNames("COAD")## [1] "years_to_birth"
## [2] "vital_status"
## [3] "days_to_death"
## [4] "days_to_last_followup"
## [5] "tumor_tissue_site"
## [6] "pathologic_stage"
## [7] "pathology_T_stage"
## [8] "pathology_N_stage"
## [9] "pathology_M_stage"
## [10] "gender"
## [11] "date_of_initial_pathologic_diagnosis"
## [12] "days_to_last_known_alive"
## [13] "radiation_therapy"
## [14] "histological_type"
## [15] "residual_tumor"
## [16] "number_of_lymph_nodes"
## [17] "race"
## [18] "ethnicity"Warning: some names may not exactly match the
colData names in the object due to differences in variable
types. These variables are kept separate and differentiated with
x and y. For example,
vital_status in this case corresponds to two different
variables obtained from the pipeline. One variable is interger type and
the other character:
## [1] "integer"## [1] "character"##
## 0 1
## 355 102##
## DECEASED LIVING
## 22 179Such conflicts should be inspected in this manner, and conflicts resolved by choosing the more complete variable, or by treating any conflicting values as unknown (“NA”).
Converting Assays to SummarizedExperiment
This section gives an overview of the operations that can be
performed on a given set of metadata obtained particularly from
data-rich objects such as those obtained from
curatedTCGAData. There are several operations that work
with microRNA, methylation, mutation, and assays that have gene symbol
annotations.
CpGtoRanges
Using the methylation annotations in
IlluminaHumanMethylation450kanno.ilmn12.hg19 and the
minfi package, we look up CpG probes and convert to genomic
coordinates with CpGtoRanges. The function provides two
assays, one with mapped probes and the other with unmapped probes.
Excluding unmapped probes can be done by setting the
unmapped argument to FALSE. This will run for
both types of methylation data (27k and 450k).
methcoad <- CpGtoRanges(coad)## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 535 sampleMap rows not in names(experiments)
qreduceTCGA
The qreduceTCGA function converts
RaggedExperiment mutation data objects to
RangedSummarizedExperiment using org.Hs.eg.db
and the qreduceTCGA utility function from
RaggedExperiment to summarize ‘silent’ and ‘non-silent’
mutations based on a ‘Variant_Classification’ metadata column in the
original object.
It uses ‘hg19’ transcript database (‘TxDb’) package internally to
summarize regions using qreduceAssay. The current genome
build (‘hg18’) in the data must be translated to ‘hg19’.
In this example, we first set the appropriate build name in the
mutation dataset COAD_Mutation-20160128 according to the NCBI
website and we then use seqlevelsStyle to match the
UCSC style in the chain.
rag <- "COAD_Mutation-20160128"
# add the appropriate genome annotation
genome(coad[[rag]]) <- "NCBI36"
# change the style to UCSC
seqlevelsStyle(rowRanges(coad[[rag]])) <- "UCSC"
# inspect changes
seqlevels(rowRanges(coad[[rag]]))## [1] "chr1" "chr2" "chr3" "chr4" "chr5" "chr6" "chr7" "chr8" "chr9"
## [10] "chr10" "chr11" "chr12" "chr13" "chr14" "chr15" "chr16" "chr17" "chr18"
## [19] "chr19" "chr20" "chr21" "chr22" "chrX" "chrY"
genome(coad[[rag]])## chr1 chr2 chr3 chr4 chr5 chr6 chr7 chr8 chr9 chr10 chr11
## "hg18" "hg18" "hg18" "hg18" "hg18" "hg18" "hg18" "hg18" "hg18" "hg18" "hg18"
## chr12 chr13 chr14 chr15 chr16 chr17 chr18 chr19 chr20 chr21 chr22
## "hg18" "hg18" "hg18" "hg18" "hg18" "hg18" "hg18" "hg18" "hg18" "hg18" "hg18"
## chrX chrY
## "hg18" "hg18"Now we use liftOver from rtracklayer to
translate ‘hg18’ builds to ‘hg19’ using the chain file obtained via
AnnotationHub. We use a query to find the file. You can
also query with terms such as
c("Homo sapiens", "chain", "hg18", "hg19") in the query
function. We are specifically looking for the chain file
“hg18ToHg19.over.chain.gz”.
library(AnnotationHub)
ah <- AnnotationHub()
query(ah, "hg18ToHg19.over.chain.gz")## AnnotationHub with 1 record
## # snapshotDate(): 2026-06-30
## # names(): AH14220
## # $dataprovider: UCSC
## # $species: Homo sapiens
## # $rdataclass: ChainFile
## # $rdatadateadded: 2014-12-15
## # $title: hg18ToHg19.over.chain.gz
## # $description: UCSC liftOver chain file from hg18 to hg19
## # $taxonomyid: 9606
## # $genome: hg18
## # $sourcetype: Chain
## # $sourceurl: http://hgdownload.cse.ucsc.edu/goldenpath/hg18/liftOver/hg18To...
## # $sourcesize: NA
## # $tags: c("liftOver", "chain", "UCSC", "genome", "homology")
## # retrieve record with 'object[["AH14220"]]'
chain <- ah[["AH14220"]]## loading from cache
ranges19 <- rtracklayer::liftOver(rowRanges(coad[[rag]]), chain)Note. The same can be done to convert
hg19 to hg38 (the same build that the Genomic
Data Commons uses) with the corresponding chain file as obtained
above.
This will give us a list of ranges, each element corresponding to a
single row in the RaggedExperiment. We remove rows that had
no matches in the liftOver process and replace the ranges
in the original RaggedExperiment with the replacement
method. Finally, we put the RaggedExperiment object back
into the MultiAssayExperiment.
re19 <- coad[[rag]][as.logical(lengths(ranges19))]
ranges19 <- unlist(ranges19)
genome(ranges19) <- "hg19"
rowRanges(re19) <- ranges19
# replacement
coad[["COAD_Mutation-20160128"]] <- re19
rowRanges(re19)## GRanges object with 62523 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr20 1552407-1552408 +
## [2] chr1 161736152-161736153 +
## [3] chr7 100685895 +
## [4] chr7 103824453 +
## [5] chr7 104783644 +
## ... ... ... ...
## [62519] chr9 36369716 +
## [62520] chr9 37692640 +
## [62521] chr9 6007456 +
## [62522] chrX 123785782 +
## [62523] chrX 51487184 +
## -------
## seqinfo: 24 sequences from hg19 genome; no seqlengthsNow that we have matching builds, we can finally run the
qreduceTCGA function.
coad <- qreduceTCGA(coad, keep.assay = TRUE)## 24 genes were dropped because they have exons located on both strands of the
## same reference sequence or on more than one reference sequence, so cannot be
## represented by a single genomic range.
## Use 'single.strand.genes.only=FALSE' to get all the genes in a GRangesList
## object, or use suppressMessages() to suppress this message.## Warning in (function (seqlevels, genome, new_style) : cannot switch some hg19's
## seqlevels from UCSC to NCBI style## ## 'select()' returned 1:1 mapping between keys and columns## Warning in .normarg_seqlevelsStyle(value): more than one seqlevels style
## supplied, using the 1st one only
## Warning in .normarg_seqlevelsStyle(value): cannot switch some hg19's seqlevels
## from UCSC to NCBI style
symbolsToRanges
In the cases where row annotations indicate gene symbols, the
symbolsToRanges utility function converts genes to genomic
ranges and replaces existing assays with
RangedSummarizedExperiment objects. Gene annotations are
given as ‘hg19’ genomic regions.
symbolsToRanges(coad)## 24 genes were dropped because they have exons located on both strands of the
## same reference sequence or on more than one reference sequence, so cannot be
## represented by a single genomic range.
## Use 'single.strand.genes.only=FALSE' to get all the genes in a GRangesList
## object, or use suppressMessages() to suppress this message.## Warning in (function (seqlevels, genome, new_style) : cannot switch some hg19's
## seqlevels from UCSC to NCBI style## 'select()' returned 1:1 mapping between keys and columns
## 24 genes were dropped because they have exons located on both strands of the
## same reference sequence or on more than one reference sequence, so cannot be
## represented by a single genomic range.
## Use 'single.strand.genes.only=FALSE' to get all the genes in a GRangesList
## object, or use suppressMessages() to suppress this message.## Warning in (function (seqlevels, genome, new_style) : cannot switch some hg19's
## seqlevels from UCSC to NCBI style## 'select()' returned 1:1 mapping between keys and columns## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 363 sampleMap rows not in names(experiments)## A MultiAssayExperiment object of 11 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 11:
## [1] COAD_CNASeq-20160128: RaggedExperiment with 40530 rows and 136 columns
## [2] COAD_miRNASeqGene-20160128: SummarizedExperiment with 705 rows and 221 columns
## [3] COAD_Mutation-20160128: RaggedExperiment with 62523 rows and 154 columns
## [4] COAD_Methylation_methyl27-20160128: SummarizedExperiment with 27578 rows and 202 columns
## [5] COAD_Methylation_methyl450-20160128: SummarizedExperiment with 485577 rows and 333 columns
## [6] COAD_Mutation-20160128_simplified: RangedSummarizedExperiment with 28617 rows and 154 columns
## [7] COAD_CNASeq-20160128_simplified: RangedSummarizedExperiment with 28617 rows and 136 columns
## [8] COAD_mRNAArray-20160128_ranged: RangedSummarizedExperiment with 14252 rows and 172 columns
## [9] COAD_mRNAArray-20160128_unranged: SummarizedExperiment with 3562 rows and 172 columns
## [10] COAD_RNASeq2GeneNorm-20160128_ranged: RangedSummarizedExperiment with 17088 rows and 191 columns
## [11] COAD_RNASeq2GeneNorm-20160128_unranged: SummarizedExperiment with 3413 rows and 191 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesImporting TCGA text data files to Bioconductor classes
A few functions in the package accept either files or classes such as
data.frame and FirehoseGISTIC as input and
return standard Bioconductor classes.
Work around for long file names on Windows
Due to file name length, Windows may not be able to read / display
all files. The workaround uses the fileNames argument from
a character vector of file names and will convert them to TCGA
barcodes.
## Load example file found in package
pkgDir <- system.file("extdata", package = "TCGAutils", mustWork = TRUE)
exonFile <- list.files(pkgDir, pattern = "cation\\.txt$", full.names = TRUE)
exonFile## [1] "/tmp/Rtmpr8lSRz/temp_libpath10642ef8e6b7/TCGAutils/extdata/bt.exon_quantification.txt"
## We add the original file prefix to query for the UUID and get the
## TCGAbarcode
filePrefix <- "unc.edu.32741f9a-9fec-441f-96b4-e504e62c5362.1755371."
## Add actual file name manually
makeGRangesListFromExonFiles(exonFile, getBarcodes = FALSE,
fileNames = paste0(filePrefix, basename(exonFile)))##
## ── Column specification ────────────────────────────────────────────────────────
## cols(
## exon = col_character(),
## raw_counts = col_double(),
## median_length_normalized = col_double(),
## RPKM = col_double()
## )## GRangesList object of length 1:
## $`unc.edu.32741f9a-9fec-441f-96b4-e504e62c5362.1755371.bt.exon_quantification.txt`
## GRanges object with 100 ranges and 3 metadata columns:
## seqnames ranges strand | raw_counts median_length_normalized
## <Rle> <IRanges> <Rle> | <numeric> <numeric>
## [1] chr1 11874-12227 + | 4 0.492918
## [2] chr1 12595-12721 + | 2 0.341270
## [3] chr1 12613-12721 + | 2 0.398148
## [4] chr1 12646-12697 + | 2 0.372549
## [5] chr1 13221-14409 + | 39 0.632997
## ... ... ... ... . ... ...
## [96] chr1 881782-881925 - | 179 1
## [97] chr1 883511-883612 - | 151 1
## [98] chr1 883870-883983 - | 155 1
## [99] chr1 886507-886618 - | 144 1
## [100] chr1 887380-887519 - | 158 1
## RPKM
## <numeric>
## [1] 0.322477
## [2] 0.449436
## [3] 0.523655
## [4] 1.097661
## [5] 0.936105
## ... ...
## [96] 35.4758
## [97] 42.2492
## [98] 38.8033
## [99] 36.6933
## [100] 32.2085
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengthsNOTE. Querying the API for legacy file names is no
longer supported. In this example, we leave the file name in rather than
translate the file name to a TCGA barcode, i.e.,
getBarcodes = FALSE. However,
filenameToBarcode is still supported for other file
types.
makeGRangesListFromCopyNumber
Other processed, genomic range-based data from TCGA data can be
imported using makeGRangesListFromCopyNumber. This
tab-delimited data file of copy number alterations from bladder
urothelial carcinoma (BLCA) was obtained from the Genomic Data
Commons and is included in TCGAUtils as an example:
grlFile <- system.file("extdata", "blca_cnaseq.txt", package = "TCGAutils")
grl <- read.table(grlFile)
head(grl)## Sample Chromosome Start End Num_Probes
## 1 TCGA-BL-A0C8-01A-11D-A10R-02 14 70362113 73912204 NA
## 2 TCGA-BL-A0C8-01A-11D-A10R-02 9 115609546 131133898 NA
## 5 TCGA-BL-A13I-01A-11D-A13U-02 13 19020028 49129100 NA
## 6 TCGA-BL-A13I-01A-11D-A13U-02 1 10208 246409808 NA
## 9 TCGA-BL-A13J-01A-11D-A10R-02 23 3119586 5636448 NA
## 10 TCGA-BL-A13J-01A-11D-A10R-02 7 10127 35776912 NA
## Segment_Mean
## 1 -0.182879931
## 2 0.039675162
## 5 0.002085552
## 6 -0.014224752
## 9 0.877072555
## 10 0.113873871
makeGRangesListFromCopyNumber(grl, split.field = "Sample")## GRangesList object of length 116:
## $`TCGA-BL-A0C8-01A-11D-A10R-02`
## GRanges object with 2 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] 14 70362113-73912204 *
## [2] 9 115609546-131133898 *
## -------
## seqinfo: 24 sequences from an unspecified genome; no seqlengths
##
## $`TCGA-BL-A13I-01A-11D-A13U-02`
## GRanges object with 2 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] 13 19020028-49129100 *
## [2] 1 10208-246409808 *
## -------
## seqinfo: 24 sequences from an unspecified genome; no seqlengths
##
## $`TCGA-BL-A13J-01A-11D-A10R-02`
## GRanges object with 2 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] 23 3119586-5636448 *
## [2] 7 10127-35776912 *
## -------
## seqinfo: 24 sequences from an unspecified genome; no seqlengths
##
## ...
## <113 more elements>
makeGRangesListFromCopyNumber(grl, split.field = "Sample",
keep.extra.columns = TRUE)## GRangesList object of length 116:
## $`TCGA-BL-A0C8-01A-11D-A10R-02`
## GRanges object with 2 ranges and 2 metadata columns:
## seqnames ranges strand | Num_Probes Segment_Mean
## <Rle> <IRanges> <Rle> | <logical> <numeric>
## [1] 14 70362113-73912204 * | <NA> -0.1828799
## [2] 9 115609546-131133898 * | <NA> 0.0396752
## -------
## seqinfo: 24 sequences from an unspecified genome; no seqlengths
##
## $`TCGA-BL-A13I-01A-11D-A13U-02`
## GRanges object with 2 ranges and 2 metadata columns:
## seqnames ranges strand | Num_Probes Segment_Mean
## <Rle> <IRanges> <Rle> | <logical> <numeric>
## [1] 13 19020028-49129100 * | <NA> 0.00208555
## [2] 1 10208-246409808 * | <NA> -0.01422475
## -------
## seqinfo: 24 sequences from an unspecified genome; no seqlengths
##
## $`TCGA-BL-A13J-01A-11D-A10R-02`
## GRanges object with 2 ranges and 2 metadata columns:
## seqnames ranges strand | Num_Probes Segment_Mean
## <Rle> <IRanges> <Rle> | <logical> <numeric>
## [1] 23 3119586-5636448 * | <NA> 0.877073
## [2] 7 10127-35776912 * | <NA> 0.113874
## -------
## seqinfo: 24 sequences from an unspecified genome; no seqlengths
##
## ...
## <113 more elements>
makeSummarizedExperimentFromGISTIC
This function is only used for converting the
FirehoseGISTIC class of the RTCGAToolbox
package. It allows the user to obtain thresholded by gene data,
probabilities and peak regions.
tempDIR <- tempdir()
co <- getFirehoseData("COAD", clinical = FALSE, GISTIC = TRUE,
destdir = tempDIR)
selectType(co, "GISTIC")## Dataset:COAD## FirehoseGISTIC object, dim: 24776 454
class(selectType(co, "GISTIC"))## [1] "FirehoseGISTIC"
## attr(,"package")
## [1] "RTCGAToolbox"
makeSummarizedExperimentFromGISTIC(co, "Peaks")## class: RangedSummarizedExperiment
## dim: 66 451
## metadata(0):
## assays(1): ''
## rownames(66): 23 24 ... 65 66
## rowData names(12): rowRanges Unique.Name ... V461 type
## colnames(451): TCGA-3L-AA1B-01A-11D-A36W-01
## TCGA-4N-A93T-01A-11D-A36W-01 ... TCGA-T9-A92H-01A-11D-A36W-01
## TCGA-WS-AB45-01A-11D-A40O-01
## colData names(0):
mergeColData: expanding the colData of a
MultiAssayExperiment
This function merges a data.frame or
DataFrame into the colData of an existing
MultiAssayExperiment object. It will match column names and
row names to do a full merge of both data sets. This convenience
function can be used, for example, to add subtype information available
for a subset of patients to the colData. Here is a
simplified example of adding a column to the colData
DataFrame:
race_df <- DataFrame(race_f = factor(colData(coad)[["race"]]),
row.names = rownames(colData(coad)))
mergeColData(coad, race_df)## A MultiAssayExperiment object of 9 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 9:
## [1] COAD_CNASeq-20160128: RaggedExperiment with 40530 rows and 136 columns
## [2] COAD_miRNASeqGene-20160128: SummarizedExperiment with 705 rows and 221 columns
## [3] COAD_mRNAArray-20160128: SummarizedExperiment with 17814 rows and 172 columns
## [4] COAD_Mutation-20160128: RaggedExperiment with 62523 rows and 154 columns
## [5] COAD_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 191 columns
## [6] COAD_Methylation_methyl27-20160128: SummarizedExperiment with 27578 rows and 202 columns
## [7] COAD_Methylation_methyl450-20160128: SummarizedExperiment with 485577 rows and 333 columns
## [8] COAD_Mutation-20160128_simplified: RangedSummarizedExperiment with 28617 rows and 154 columns
## [9] COAD_CNASeq-20160128_simplified: RangedSummarizedExperiment with 28617 rows and 136 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesTranslating and interpreting TCGA identifiers
GDC Data Updates
Data Release (version 32.0) changed the behavior of the ID translation functions. This is due to the removal of files whose UUIDs were translated. These files have been replaced with newer runs of the pipeline. The new UUIDs can be mapped to the old UUIDs with project specific maps located on GitHub at https://github.com/NCI-GDC/gdc-docs/tree/develop/docs/Data/Release_Notes/GCv36_Manifests as well as with the history lookup API endpoint.
Note. The GDC API version v3.28.0
has deprecated the /legacy/files endpoint; therefore,
legacy data is no longer available.
UUID History Lookup
Facilities in the GDC API allow one to look up old UUIDs and obtain
new ones. The UUIDhistory function returns a
data.frame with old and new UUIDs for a single UUID lookup.
This will allow one to map old UUIDs with new ones in order to run a
translation query. Old UUIDs have been removed from the API and can no
longer be translated by the functions provided in this package.
UUIDhistory("0001801b-54b0-4551-8d7a-d66fb59429bf")## uuid version file_change release_date
## 1 0001801b-54b0-4551-8d7a-d66fb59429bf 1 superseded 2018-08-23
## 2 b4bce3ff-7fdc-4849-880b-56f2b348ceac 2 released 2022-03-29
## data_release
## 1 12.0
## 2 32.0Translation
The TCGA project has generated massive amounts of data. Some data can
be obtained with Universally Unique
IDentifiers (UUID) and other data with
TCGA barcodes. The Genomic Data Commons provides a JSON API for mapping
between UUID and barcode, but it is difficult for many people to
understand. TCGAutils makes simple functions available for
two-way translation between vectors of these identifiers.
TCGA barcode to UUID
Here we translate the first two TCGA barcodes of the previous copy-number alterations dataset to UUID:
## [1] "TCGA-A6-2671-01A-01D-1405-02" "TCGA-A6-2671-10A-01D-1405-02"
## [3] "TCGA-A6-2674-01A-02D-1167-02" "TCGA-A6-2674-10A-01D-1167-02"
barcodeToUUID(xbarcode)## submitter_aliquot_ids aliquot_ids
## 46 TCGA-A6-2671-01A-01D-1405-02 82e23baf-da11-4175-bee0-81c0c0137d72
## 70 TCGA-A6-2671-10A-01D-1405-02 da65c9d3-62ac-4fb5-b452-1e9c551ba243
## 11 TCGA-A6-2674-01A-02D-1167-02 9fdfc199-b878-4049-994e-b5ca384678fb
## 38 TCGA-A6-2674-10A-01D-1167-02 dd75656d-6df6-4c53-972d-439791c908acUUID to TCGA barcode
Here we have a known case UUID that we want to translate into a TCGA barcode.
UUIDtoBarcode("ae55b2d3-62a1-419e-9f9a-5ddfac356db4", from_type = "case_id")## case_id submitter_id
## 1 ae55b2d3-62a1-419e-9f9a-5ddfac356db4 TCGA-B0-5117In cases where we want to translate a known file UUID to the
associated TCGA patient barcode, we can use
UUIDtoBarcode.
UUIDtoBarcode("b4bce3ff-7fdc-4849-880b-56f2b348ceac", from_type = "file_id")## file_id associated_entities.entity_submitter_id
## 1 b4bce3ff-7fdc-4849-880b-56f2b348ceac TCGA-B0-5094-11A-01D-1421-08Translating aliquot UUIDs is also possible by providing a known
aliquot UUID to the function and giving a from_type,
“aliquot_ids”:
UUIDtoBarcode("d85d8a17-8aea-49d3-8a03-8f13141c163b", from_type = "aliquot_ids")## portions.analytes.aliquots.aliquot_id portions.analytes.aliquots.submitter_id
## 1 d85d8a17-8aea-49d3-8a03-8f13141c163b TCGA-CV-5443-01A-01D-1510-01Additional UUIDs may be supported in future versions.
UUID to UUID
We can also translate from file UUIDs to case UUIDs and vice versa as
long as we know the input type. We can use the case UUID from the
previous example to get the associated file UUIDs using
UUIDtoUUID. Note that this translation is a one to many
relationship, thus yielding a data.frame of file UUIDs for
a single case UUID.
head(UUIDtoUUID("ae55b2d3-62a1-419e-9f9a-5ddfac356db4", to_type = "file_id"))## case_id files.file_id
## 1 ae55b2d3-62a1-419e-9f9a-5ddfac356db4 3d9a866a-7bb3-4cdc-8e77-6263745f2e74
## 2 ae55b2d3-62a1-419e-9f9a-5ddfac356db4 e20b7e0e-fa86-43d8-af3c-6cefdec05ae2
## 3 ae55b2d3-62a1-419e-9f9a-5ddfac356db4 b45f91db-13da-4ce2-9cfa-df97e73c9138
## 4 ae55b2d3-62a1-419e-9f9a-5ddfac356db4 30a79c48-9bae-41e2-9865-1a6b1ef9dab1
## 5 ae55b2d3-62a1-419e-9f9a-5ddfac356db4 ba07ad3d-5e92-4091-a853-5f9e49327378
## 6 ae55b2d3-62a1-419e-9f9a-5ddfac356db4 c0669db0-a20c-4836-aca7-0deb6ae8ba0cOne possible way to verify that file IDs are matching case UUIDS is
to browse to the Genomic Data Commons webpage with the specific file
UUID. Here we look at the first file UUID entry in the output
data.frame:
https://portal.gdc.cancer.gov/files/0ff55a5e-6058-4e0b-9641-e3cb375ff214
In the page we check that the case UUID matches the input.
Parsing TCGA barcodes
Several functions exist for working with TCGA barcodes, the main
function being TCGAbarcode. It takes a TCGA barcode and
returns information about participant, sample, and/or portion.
## Return participant barcodes
TCGAbarcode(xbarcode, participant = TRUE)## [1] "TCGA-A6-2671" "TCGA-A6-2671" "TCGA-A6-2674" "TCGA-A6-2674"
## Just return samples
TCGAbarcode(xbarcode, participant = FALSE, sample = TRUE)## [1] "01A" "10A" "01A" "10A"
## Include sample data as well
TCGAbarcode(xbarcode, participant = TRUE, sample = TRUE)## [1] "TCGA-A6-2671-01A" "TCGA-A6-2671-10A" "TCGA-A6-2674-01A" "TCGA-A6-2674-10A"
## Include portion and analyte data
TCGAbarcode(xbarcode, participant = TRUE, sample = TRUE, portion = TRUE)## [1] "TCGA-A6-2671-01A-01D" "TCGA-A6-2671-10A-01D" "TCGA-A6-2674-01A-02D"
## [4] "TCGA-A6-2674-10A-01D"Sample selection
Based on lookup table values, the user can select certain sample types from a vector of sample barcodes. Below we select “Primary Solid Tumors” from a vector of barcodes, returning a logical vector identifying the matching samples.
## Select primary solid tumors
TCGAsampleSelect(xbarcode, "01")## 01 10 01 10
## TRUE FALSE TRUE FALSE
## Select blood derived normals
TCGAsampleSelect(xbarcode, "10")## 01 10 01 10
## FALSE TRUE FALSE TRUEPrimary tumors
We provide a TCGAprimaryTumors helper function to
facilitate the selection of primary tumor samples only:
TCGAprimaryTumors(coad)## harmonizing input:
## removing 233 sampleMap rows with 'colname' not in colnames of experiments## A MultiAssayExperiment object of 9 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 9:
## [1] COAD_CNASeq-20160128: RaggedExperiment with 40530 rows and 68 columns
## [2] COAD_miRNASeqGene-20160128: SummarizedExperiment with 705 rows and 220 columns
## [3] COAD_mRNAArray-20160128: SummarizedExperiment with 17814 rows and 153 columns
## [4] COAD_Mutation-20160128: RaggedExperiment with 62523 rows and 154 columns
## [5] COAD_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 191 columns
## [6] COAD_Methylation_methyl27-20160128: SummarizedExperiment with 27578 rows and 165 columns
## [7] COAD_Methylation_methyl450-20160128: SummarizedExperiment with 485577 rows and 293 columns
## [8] COAD_Mutation-20160128_simplified: RangedSummarizedExperiment with 28617 rows and 154 columns
## [9] COAD_CNASeq-20160128_simplified: RangedSummarizedExperiment with 28617 rows and 68 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat files
data.frame representation of barcode
The straightforward TCGAbiospec function will take the
information contained in the TCGA barcode and display it in
data.frame format with appropriate column names.
TCGAbiospec(xbarcode)## submitter_id sample_definition sample vial portion analyte plate center
## 1 TCGA-A6-2671 Primary Solid Tumor 01 A 01 D 1405 02
## 2 TCGA-A6-2671 Blood Derived Normal 10 A 01 D 1405 02
## 3 TCGA-A6-2674 Primary Solid Tumor 01 A 02 D 1167 02
## 4 TCGA-A6-2674 Blood Derived Normal 10 A 01 D 1167 02OncoPrint - oncoPrintTCGA
We provide a convenience function that investigates metadata within
curatedTCGAData objects to present a plot of molecular
alterations within a paricular cancer. MultiAssayExperiment
objects are required to have an identifiable ‘Mutation’ assay
(using text search). The variantCol argument identifies the
mutation type column within the data.
Note. Functionality streamlined from the
ComplexHeatmap package.
oncoPrintTCGA(coad, matchassay = rag)## 24 genes were dropped because they have exons located on both strands of the
## same reference sequence or on more than one reference sequence, so cannot be
## represented by a single genomic range.
## Use 'single.strand.genes.only=FALSE' to get all the genes in a GRangesList
## object, or use suppressMessages() to suppress this message.## Warning in (function (seqlevels, genome, new_style) : cannot switch some hg19's
## seqlevels from UCSC to NCBI style## 'select()' returned 1:1 mapping between keys and columns## All mutation types: Frame Shift Del, Frame Shift Ins, Intron, Missense
## Mutation, Nonsense Mutation.## `alter_fun` is assumed vectorizable. If it does not generate correct
## plot, please set `alter_fun_is_vectorized = FALSE` in `oncoPrint()`.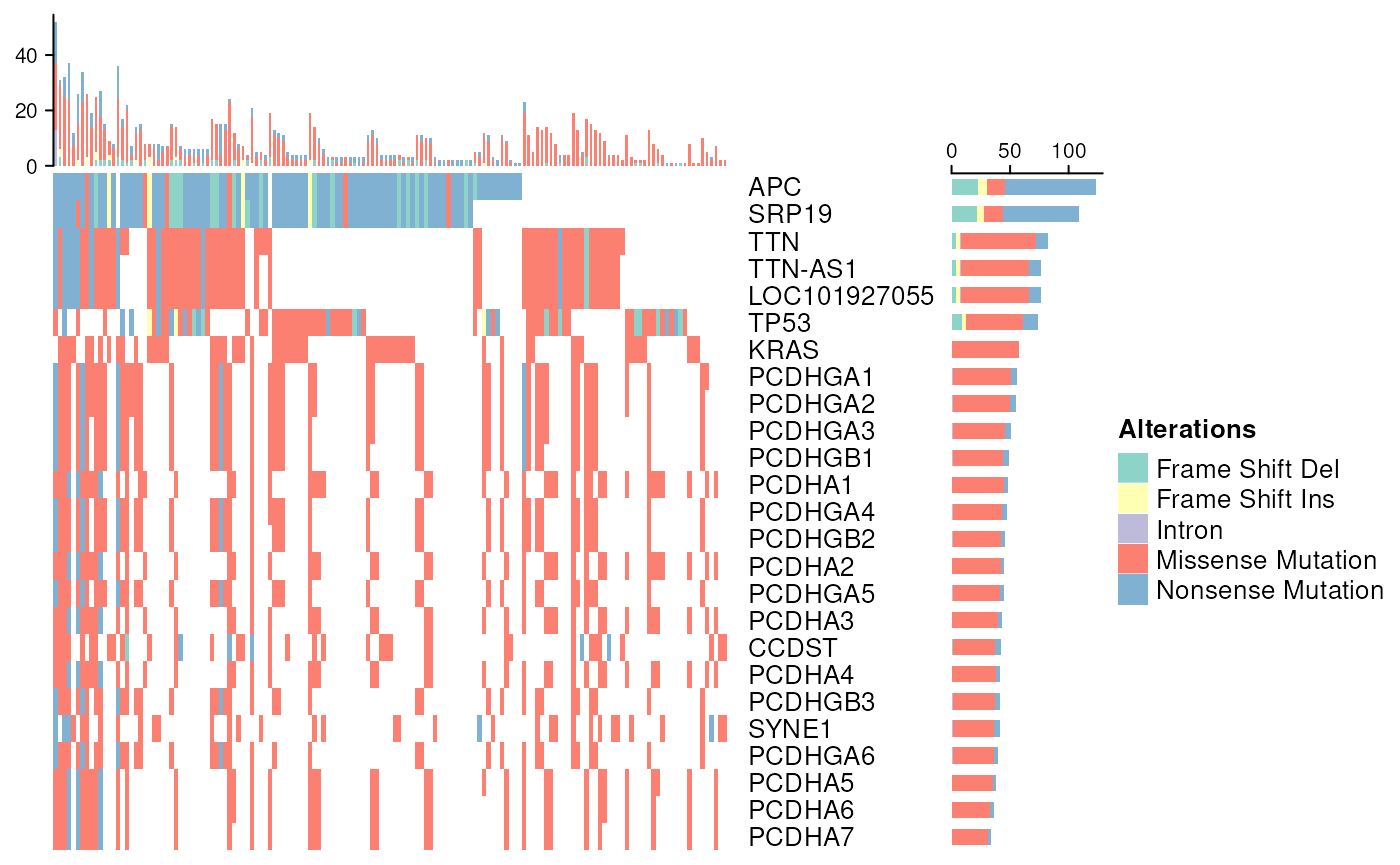
Reference data
The TCGAutils package provides several helper datasets
for working with TCGA barcodes.
sampleTypes
As shown previously, the reference dataset sampleTypes
defines sample codes and their sample types (see
?sampleTypes for source url).
## Obtained previously
sampleCodes <- TCGAbarcode(xbarcode, participant = FALSE, sample = TRUE)
## Lookup table
head(sampleTypes)## Code Definition Short.Letter.Code
## 1 01 Primary Solid Tumor TP
## 2 02 Recurrent Solid Tumor TR
## 3 03 Primary Blood Derived Cancer - Peripheral Blood TB
## 4 04 Recurrent Blood Derived Cancer - Bone Marrow TRBM
## 5 05 Additional - New Primary TAP
## 6 06 Metastatic TM
## Match codes found in the barcode to the lookup table
sampleTypes[match(unique(substr(sampleCodes, 1L, 2L)), sampleTypes[["Code"]]), ]## Code Definition Short.Letter.Code
## 1 01 Primary Solid Tumor TP
## 10 10 Blood Derived Normal NBSource: https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/sample-type-codes
clinicalNames - Firehose pipeline clinical
variables
clinicalNames is a list of the level 4 variable names
(the most commonly used clinical and pathological variables, with
follow-ups merged) from each colData datasets in
curatedTCGAData. Shipped curatedTCGAData
MultiAssayExperiment objects merge additional levels 1-3
clinical, pathological, and biospecimen data and contain many more
variables than the ones listed here.
data("clinicalNames")
clinicalNames## CharacterList of length 33
## [["ACC"]] years_to_birth vital_status days_to_death ... race ethnicity
## [["BLCA"]] years_to_birth vital_status days_to_death ... race ethnicity
## [["BRCA"]] years_to_birth vital_status days_to_death ... race ethnicity
## [["CESC"]] years_to_birth vital_status ... age_at_diagnosis clinical_stage
## [["CHOL"]] years_to_birth vital_status days_to_death ... race ethnicity
## [["COAD"]] years_to_birth vital_status days_to_death ... race ethnicity
## [["DLBC"]] years_to_birth vital_status days_to_death ... race ethnicity
## [["ESCA"]] years_to_birth vital_status days_to_death ... race ethnicity
## [["GBM"]] years_to_birth vital_status days_to_death ... race ethnicity
## [["HNSC"]] years_to_birth vital_status days_to_death ... race ethnicity
## ...
## <23 more elements>
lengths(clinicalNames)## ACC BLCA BRCA CESC CHOL COAD DLBC ESCA GBM HNSC KICH KIRC KIRP LAML LGG LIHC
## 16 18 17 48 16 18 11 19 12 19 18 19 19 9 12 16
## LUAD LUSC MESO OV PAAD PCPG PRAD READ SARC SKCM STAD TGCT THCA THYM UCEC UCS
## 20 20 17 12 19 13 18 18 12 17 17 15 21 11 9 11
## UVM
## 14
sessionInfo
## R version 4.6.0 (2026-04-24)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: Etc/UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] AnnotationHub_4.3.2 BiocFileCache_3.3.0
## [3] dbplyr_2.6.0 rhdf5_2.57.1
## [5] RaggedExperiment_1.37.0 GenomeInfoDb_1.49.1
## [7] R.utils_2.13.0 R.oo_1.27.1
## [9] R.methodsS3_1.8.2 rtracklayer_1.73.0
## [11] RTCGAToolbox_2.43.0 curatedTCGAData_1.35.0
## [13] MultiAssayExperiment_1.39.0 SummarizedExperiment_1.43.0
## [15] Biobase_2.73.1 GenomicRanges_1.65.0
## [17] Seqinfo_1.3.0 IRanges_2.47.2
## [19] S4Vectors_0.51.5 BiocGenerics_0.59.8
## [21] generics_0.1.4 MatrixGenerics_1.25.0
## [23] matrixStats_1.5.0 TCGAutils_1.32.2
## [25] BiocStyle_2.41.0
##
## loaded via a namespace (and not attached):
## [1] RColorBrewer_1.1-3
## [2] shape_1.4.6.1
## [3] jsonlite_2.0.0
## [4] magrittr_2.0.5
## [5] GenomicFeatures_1.65.0
## [6] rmarkdown_2.31
## [7] GlobalOptions_0.1.4
## [8] fs_2.1.0
## [9] BiocIO_1.23.3
## [10] ragg_1.5.2
## [11] vctrs_0.7.3
## [12] memoise_2.0.1
## [13] Rsamtools_2.29.0
## [14] RCurl_1.98-1.19
## [15] htmltools_0.5.9
## [16] S4Arrays_1.13.0
## [17] TxDb.Hsapiens.UCSC.hg19.knownGene_3.22.1
## [18] BiocBaseUtils_1.15.1
## [19] curl_7.1.0
## [20] Rhdf5lib_2.1.0
## [21] SparseArray_1.13.2
## [22] sass_0.4.10
## [23] bslib_0.11.0
## [24] htmlwidgets_1.6.4
## [25] desc_1.4.3
## [26] httr2_1.2.3
## [27] cachem_1.1.0
## [28] GenomicAlignments_1.49.0
## [29] lifecycle_1.0.5
## [30] iterators_1.0.14
## [31] pkgconfig_2.0.3
## [32] Matrix_1.7-5
## [33] R6_2.6.1
## [34] fastmap_1.2.0
## [35] clue_0.3-68
## [36] digest_0.6.39
## [37] colorspace_2.1-2
## [38] AnnotationDbi_1.75.0
## [39] ExperimentHub_3.3.0
## [40] textshaping_1.0.5
## [41] RSQLite_3.53.3
## [42] org.Hs.eg.db_3.23.1
## [43] filelock_1.0.3
## [44] RJSONIO_2.0.5
## [45] httr_1.4.8
## [46] abind_1.4-8
## [47] compiler_4.6.0
## [48] bit64_4.8.2
## [49] withr_3.0.3
## [50] doParallel_1.0.17
## [51] BiocParallel_1.47.0
## [52] DBI_1.3.0
## [53] HDF5Array_1.41.0
## [54] rappdirs_0.3.4
## [55] DelayedArray_0.39.3
## [56] rjson_0.2.23
## [57] tools_4.6.0
## [58] otel_0.2.0
## [59] glue_1.8.1
## [60] h5mread_1.5.0
## [61] restfulr_0.0.17
## [62] rhdf5filters_1.25.0
## [63] grid_4.6.0
## [64] cluster_2.1.8.2
## [65] tzdb_0.5.0
## [66] data.table_1.18.4
## [67] hms_1.1.4
## [68] xml2_1.6.0
## [69] XVector_0.53.0
## [70] BiocVersion_3.24.0
## [71] foreach_1.5.2
## [72] pillar_1.11.1
## [73] stringr_1.6.0
## [74] circlize_0.4.18
## [75] dplyr_1.2.1
## [76] lattice_0.22-9
## [77] bit_4.6.0
## [78] tidyselect_1.2.1
## [79] ComplexHeatmap_2.29.0
## [80] Biostrings_2.81.3
## [81] knitr_1.51
## [82] bookdown_0.47
## [83] xfun_0.59
## [84] stringi_1.8.7
## [85] UCSC.utils_1.9.0
## [86] yaml_2.3.12
## [87] evaluate_1.0.5
## [88] codetools_0.2-20
## [89] cigarillo_1.3.0
## [90] tibble_3.3.1
## [91] BiocManager_1.30.27
## [92] cli_3.6.6
## [93] systemfonts_1.3.2
## [94] jquerylib_0.1.4
## [95] GenomicDataCommons_1.37.0
## [96] png_0.1-9
## [97] XML_3.99-0.23
## [98] parallel_4.6.0
## [99] pkgdown_2.2.0
## [100] readr_2.2.0
## [101] blob_1.3.0
## [102] bitops_1.0-9
## [103] purrr_1.2.2
## [104] crayon_1.5.3
## [105] GetoptLong_1.1.1
## [106] rlang_1.2.0
## [107] KEGGREST_1.53.1
## [108] rvest_1.0.5