MultiAssayExperiment: The Integrative Bioconductor Container
MultiAssay Special Interest Group
March 28, 2026
Source:vignettes/MultiAssayExperiment.Rmd
MultiAssayExperiment.RmdInstallation
if (!require("BiocManager"))
install.packages("BiocManager")
BiocManager::install("MultiAssayExperiment")Loading the packages:
Citing MultiAssayExperiment
Without your citations our free and open-source software would not be
possible. Please cite MultiAssayExperiment as shown in the
References section (Ramos et al. (2017)). You may also refer to the
Cancer Research publication at the AACR Journals link here.
A Brief Description
MultiAssayExperiment offers a data structure for
representing and analyzing multi-omics experiments: a biological
analysis approach utilizing multiple types of observations, such as DNA
mutations and abundance of RNA and proteins, in the same biological
specimens.
Overview of the MultiAssayExperiment class
Here is an overview of the class and its constructors and extractors:
empty <- MultiAssayExperiment()
empty## A MultiAssayExperiment object of 0 listed
## experiments with no user-defined names and respective classes.
## Containing an ExperimentList class object of length 0:
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat files
slotNames(empty)## [1] "ExperimentList" "colData" "sampleMap" "drops"
## [5] "metadata"A visual representation of the MultiAssayExperiment class and its accessor functions can be seen below. There are three main components:
ExperimentListcolDatasampleMap
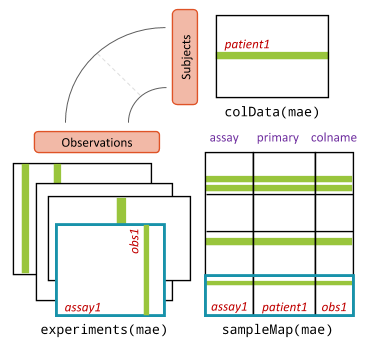
MultiAssayExperiment object schematic shows the design of the infrastructure class. The colData provides data about the patients, cell lines, or other biological units, with one row per unit and one column per variable. The experiments are a list of assay datasets of arbitrary class, with one column per observation. The sampleMap links a single table of patient data (colData) to a list of experiments via a simple but powerful table of experiment:patient edges (relationships), that can be created automatically in simple cases or in a spreadsheet if assay-specific sample identifiers are used. sampleMap relates each column (observation) in the assays (experiments) to exactly one row (biological unit) in colData; however, one row of colData may map to zero, one, or more columns per assay, allowing for missing and replicate assays. Green stripes indicate a mapping of one subject to multiple observations across experiments.
Components of the MultiAssayExperiment
ExperimentList: experimental data
The ExperimentList slot and class is the container
workhorse for the MultiAssayExperiment class. It contains
all the experimental data. It inherits from class
S4Vectors::SimpleList with one element/component per data
type.
class(experiments(empty)) # ExperimentList## [1] "ExperimentList"
## attr(,"package")
## [1] "MultiAssayExperiment"The elements of the ExperimentList can contain
ID-based and range-based data.
Requirements for all classes in the ExperimentList are
listed in the API.
The following base and Bioconductor classes are known to work as elements of the ExperimentList:
base::matrix: the base class, can be used for ID-based datasets such as gene expression summarized per-gene, microRNA, metabolomics, or microbiome data.SummarizedExperiment::SummarizedExperiment: A richer representation compared to a ordinary matrix of ID-based datasets capable of storing additional assay- level metadata.Biobase::ExpressionSet: A legacy representation of ID-based datasets, supported for convenience and supplanted bySummarizedExperiment.SummarizedExperiment::RangedSummarizedExperiment: For rectangular range-based datasets, one set of genomic ranges are assayed for multiple samples. It can be used for gene expression, methylation, or other data types that refer to genomic positions.RaggedExperiment::RaggedExperiment: For range-based datasets, such as copy number and mutation data, theRaggedExperimentclass can be used to represent measurements by genomic positions.
Class requirements within ExperimentList container
See the API
section for details on requirements for using other data classes. In
general, data classes meeting minimum requirements, including support
for square bracket [ subsetting and dimnames()
will work by default.
The datasets contained in elements of the ExperimentList
can have:
- column names (required)
- row names (optional)
The column names correspond to samples, and are used to match assay
data to specimen metadata stored in colData.
The row names can correspond to a variety of features in the data including but not limited to gene names, probe IDs, proteins, and named ranges. Note that the existence of “row” names does not mean the data must be rectangular or matrix-like.
Classes contained in the ExperimentList must support the
following list of methods:
-
[: single square bracket subsetting, with a single comma. It is assumed that values before the comma subset rows, and values after the comma subset columns. -
dimnames(): corresponding to features (such as genes, proteins, etc.) and experimental samples -
dim(): returns a vector of the number of rows and number of columns
colData: primary data
The MultiAssayExperiment keeps one set of “primary”
metadata that describes the ‘biological unit’ which can refer to
specimens, experimental subjects, patients, etc. In this vignette, we
will refer to each experimental subject as a patient.
colData slot requirements
The colData dataset should be of class
DataFrame but can accept a data.frame class
object that will be coerced.
In order to relate metadata of the biological unit, the row names of
the colData dataset must contain patient identifiers.
patient.data <- data.frame(sex=c("M", "F", "M", "F"),
age=38:41,
row.names=c("Jack", "Jill", "Bob", "Barbara"))
patient.data## sex age
## Jack M 38
## Jill F 39
## Bob M 40
## Barbara F 41Key points:
- one row of
colDatacan map to zero, one, or more columns in anyExperimentListelement - each row of
colDatamust map to at least one column in at least oneExperimentListelement. - each column of each
ExperimentListelement must map to exactly one row ofcolData.
These relationships are defined by the sampleMap.
Note on the flexibility of the DataFrame
For many typical purposes the DataFrame and
data.frame behave equivalently; but the
Dataframe is more flexible as it allows any vector-like
data type to be stored in its columns. The flexibility of the
DataFrame permits, for example, storing multiple
dose-response values for a single cell line, even if the number of doses
and responses is not consistent across all cell lines. Doses could be
stored in one column of colData as a
SimpleList, and responses in another column, also as a
SimpleList. Or, dose-response values could be stored in a
single column of colData as a two-column matrix for each
cell line.
sampleMap: relating colData to multiple
assays
The sampleMap is a DataFrame that relates
the “primary” data (colData) to the experimental
assays:
## [1] TRUEThe sampleMap provides an unambiguous map from every
experimental observation to one and only one row in
colData. It is, however, permissible for a row of
colData to be associated with multiple experimental
observations or no observations at all. In other words, there is a
“many-to-one” mapping from experimental observations to rows of
colData, and a “one-to-any-number” mapping from rows of
colData to experimental observations.
sampleMap structure
The sampleMap has three columns, with the following
column names:
assay provides the names of the different experiments / assays performed. These are user-defined, with the only requirement that the names of the
ExperimentList, where the experimental assays are stored, must be contained in this column.primary provides the “primary” sample names. All values in this column must also be present in the rownames of
colData(MultiAssayExperiment). In this example, allowable values in this column are “Jack”, “Jill”, “Barbara”, and “Bob”.colname provides the sample names used by experimental datasets, which in practice are often different than the primary sample names. For each assay, all column names must be found in this column. Otherwise, those assays would be orphaned: it would be impossible to match them up to samples in the overall experiment. As mentioned above, duplicate values are allowed, to represent replicates with the same overall experiment-level annotation.
This design is motivated by the following situations:
- It allows flexibility for any amount of technical replication and biological replication (such as tumor and matched normal for a single patient) of individual assays.
- It allows missing observations (such as RNA-seq performed only for some of the patients).
- It allows the use of different identifiers to be used for patients / specimens and for each assay. These different identifiers are matched unambiguously, and consistency between them is maintained during subsetting and re-ordering.
Instances where sampleMap isn’t provided
If each assay uses the same colnames (i.e., if the same sample
identifiers are used for each experiment), a simple list of these
datasets is sufficient for the MultiAssayExperiment
constructor function. It is not necessary for them to have the same
rownames or colnames:
exprss1 <- matrix(rnorm(16), ncol = 4,
dimnames = list(sprintf("ENST00000%i", sample(288754:290000, 4)),
c("Jack", "Jill", "Bob", "Bobby")))
exprss2 <- matrix(rnorm(12), ncol = 3,
dimnames = list(sprintf("ENST00000%i", sample(288754:290000, 4)),
c("Jack", "Jane", "Bob")))
doubleExp <- list("methyl 2k" = exprss1, "methyl 3k" = exprss2)
simpleMultiAssay <- MultiAssayExperiment(experiments=doubleExp)
simpleMultiAssay## A MultiAssayExperiment object of 2 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 2:
## [1] methyl 2k: matrix with 4 rows and 4 columns
## [2] methyl 3k: matrix with 4 rows and 3 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesIn the above example, the user did not provide the
colData argument so the constructor function filled it with
an empty DataFrame:
colData(simpleMultiAssay)## DataFrame with 5 rows and 0 columnsBut the colData can be provided. Here, note that any
assay sample (column) that cannot be mapped to a corresponding row in
the provided colData gets dropped. This is part of ensuring
internal validity of the MultiAssayExperiment.
simpleMultiAssay2 <- MultiAssayExperiment(experiments=doubleExp,
colData=patient.data)## Warning: Data dropped from ExperimentList (element - column):
## methyl 2k - Bobby
## methyl 3k - Jane
## Unable to map to rows of colData## harmonizing input:
## removing 1 colData rownames not in sampleMap 'primary'
simpleMultiAssay2## A MultiAssayExperiment object of 2 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 2:
## [1] methyl 2k: matrix with 4 rows and 3 columns
## [2] methyl 3k: matrix with 4 rows and 2 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat files
colData(simpleMultiAssay2)## DataFrame with 3 rows and 2 columns
## sex age
## <character> <integer>
## Jack M 38
## Jill F 39
## Bob M 40metadata
Metadata can be added at different levels of the
MultiAssayExperiment.
Can be of ANY class, for storing study-wide metadata, such
as citation information. For an empty MultiAssayExperiment
object, it is NULL.
## [1] "list"At the ExperimentList level, the metadata
function would allow the user to enter metadata as a
list.
metadata(experiments(empty))## list()At the individual assay level, certain classes may support
metadata, for example, metadata and mcols for
a SummarizedExperiment. It is recommended to use
metadata at the ExperimentList level.
Creating a MultiAssayExperiment object: a rich
example
In this section we demonstrate all core supported data classes, using
different sample ID conventions for each assay, with primary
colData. The some supported classes such as,
matrix, SummarizedExperiment, and
RangedSummarizedExperiment.
Create toy datasets demonstrating all supported data types
We have three matrix-like datasets. First, let’s represent expression
data as a SummarizedExperiment:
(arraydat <- matrix(seq(101, 108), ncol=4,
dimnames=list(c("ENST00000294241", "ENST00000355076"),
c("array1", "array2", "array3", "array4"))))## array1 array2 array3 array4
## ENST00000294241 101 103 105 107
## ENST00000355076 102 104 106 108
coldat <- data.frame(slope53=rnorm(4),
row.names=c("array1", "array2", "array3", "array4"))
exprdat <- SummarizedExperiment(arraydat, colData=coldat)
exprdat## class: SummarizedExperiment
## dim: 2 4
## metadata(0):
## assays(1): ''
## rownames(2): ENST00000294241 ENST00000355076
## rowData names(0):
## colnames(4): array1 array2 array3 array4
## colData names(1): slope53The following map matches colData sample names to
exprdata sample names. Note that row orders aren’t
initially matched up, and this is OK.
(exprmap <- data.frame(primary=rownames(patient.data)[c(1, 2, 4, 3)],
colname=c("array1", "array2", "array3", "array4"),
stringsAsFactors = FALSE))## primary colname
## 1 Jack array1
## 2 Jill array2
## 3 Barbara array3
## 4 Bob array4Now methylation data, which we will represent as a
matrix. It uses gene identifiers also, but measures a
partially overlapping set of genes. Now, let’s store this as a simple
matrix which can contains a replicate for one of the patients.
(methyldat <-
matrix(1:10, ncol=5,
dimnames=list(c("ENST00000355076", "ENST00000383706"),
c("methyl1", "methyl2", "methyl3",
"methyl4", "methyl5"))))## methyl1 methyl2 methyl3 methyl4 methyl5
## ENST00000355076 1 3 5 7 9
## ENST00000383706 2 4 6 8 10The following map matches colData sample names to
methyldat sample names.
(methylmap <- data.frame(primary = c("Jack", "Jack", "Jill", "Barbara", "Bob"),
colname = c("methyl1", "methyl2", "methyl3", "methyl4", "methyl5"),
stringsAsFactors = FALSE))## primary colname
## 1 Jack methyl1
## 2 Jack methyl2
## 3 Jill methyl3
## 4 Barbara methyl4
## 5 Bob methyl5Now we have a microRNA platform, which has no common identifiers with
the other datasets, and which we also represent as a
matrix. It is also missing data for “Jill”. We will use the
same sample naming convention as we did for arrays.
(microdat <- matrix(201:212, ncol=3,
dimnames=list(c("hsa-miR-21", "hsa-miR-191",
"hsa-miR-148a", "hsa-miR148b"),
c("micro1", "micro2", "micro3"))))## micro1 micro2 micro3
## hsa-miR-21 201 205 209
## hsa-miR-191 202 206 210
## hsa-miR-148a 203 207 211
## hsa-miR148b 204 208 212And the following map matches colData sample names to
microdat sample names.
(micromap <- data.frame(primary = c("Jack", "Barbara", "Bob"),
colname = c("micro1", "micro2", "micro3"), stringsAsFactors = FALSE))## primary colname
## 1 Jack micro1
## 2 Barbara micro2
## 3 Bob micro3Finally, we create a dataset of class
RangedSummarizedExperiment:
nrows <- 5
ncols <- 4
counts <- matrix(runif(nrows * ncols, 1, 1e4), nrows)
rowRanges <- GRanges(rep(c("chr1", "chr2"), c(2, nrows - 2)),
IRanges(floor(runif(nrows, 1e5, 1e6)), width=100),
strand=sample(c("+", "-"), nrows, TRUE),
feature_id=sprintf("ID\\%03d", 1:nrows))
names(rowRanges) <- letters[1:5]
colData <- DataFrame(Treatment=rep(c("ChIP", "Input"), 2),
row.names= c("mysnparray1", "mysnparray2", "mysnparray3", "mysnparray4"))
rse <- SummarizedExperiment(assays=SimpleList(counts=counts),
rowRanges=rowRanges, colData=colData)And we map the colData samples to the
RangedSummarizedExperiment:
(rangemap <-
data.frame(primary = c("Jack", "Jill", "Bob", "Barbara"),
colname = c("mysnparray1", "mysnparray2", "mysnparray3", "mysnparray4"),
stringsAsFactors = FALSE))## primary colname
## 1 Jack mysnparray1
## 2 Jill mysnparray2
## 3 Bob mysnparray3
## 4 Barbara mysnparray4
sampleMap creation
The MultiAssayExperiment constructor function can create
the sampleMap automatically if a single naming convention
is used, but in this example it cannot because we used platform-specific
sample identifiers (e.g. mysnparray1, etc). So we must provide an ID map
that matches the samples of each experiment back to the
colData, as a three-column data.frame or
DataFrame with three columns named “assay”, primary”, and
“colname”. Here we start with a list:
listmap <- list(exprmap, methylmap, micromap, rangemap)
names(listmap) <- c("Affy", "Methyl 450k", "Mirna", "CNV gistic")
listmap## $Affy
## primary colname
## 1 Jack array1
## 2 Jill array2
## 3 Barbara array3
## 4 Bob array4
##
## $`Methyl 450k`
## primary colname
## 1 Jack methyl1
## 2 Jack methyl2
## 3 Jill methyl3
## 4 Barbara methyl4
## 5 Bob methyl5
##
## $Mirna
## primary colname
## 1 Jack micro1
## 2 Barbara micro2
## 3 Bob micro3
##
## $`CNV gistic`
## primary colname
## 1 Jack mysnparray1
## 2 Jill mysnparray2
## 3 Bob mysnparray3
## 4 Barbara mysnparray4and use the convenience function listToMap to convert
the list of data.frame objects to a valid object for the
sampleMap:
dfmap <- listToMap(listmap)
dfmap## DataFrame with 16 rows and 3 columns
## assay primary colname
## <factor> <character> <character>
## 1 Affy Jack array1
## 2 Affy Jill array2
## 3 Affy Barbara array3
## 4 Affy Bob array4
## 5 Methyl 450k Jack methyl1
## ... ... ... ...
## 12 Mirna Bob micro3
## 13 CNV gistic Jack mysnparray1
## 14 CNV gistic Jill mysnparray2
## 15 CNV gistic Bob mysnparray3
## 16 CNV gistic Barbara mysnparray4Note, dfmap can be reverted to a list with another
provided function:
mapToList(dfmap, "assay")Experimental data as a list()
Create an named list of experiments for the
MultiAssayExperiment function. All of these names must be
found within in the third column of dfmap:
objlist <- list("Affy" = exprdat, "Methyl 450k" = methyldat,
"Mirna" = microdat, "CNV gistic" = rse)Creation of the MultiAssayExperiment class object
We recommend using the MultiAssayExperiment constructor
function:
myMultiAssay <- MultiAssayExperiment(objlist, patient.data, dfmap)
myMultiAssay## A MultiAssayExperiment object of 4 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 2 rows and 4 columns
## [2] Methyl 450k: matrix with 2 rows and 5 columns
## [3] Mirna: matrix with 4 rows and 3 columns
## [4] CNV gistic: RangedSummarizedExperiment with 5 rows and 4 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesThe following extractor functions can be used to get extract data from the object:
experiments(myMultiAssay)## ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 2 rows and 4 columns
## [2] Methyl 450k: matrix with 2 rows and 5 columns
## [3] Mirna: matrix with 4 rows and 3 columns
## [4] CNV gistic: RangedSummarizedExperiment with 5 rows and 4 columns
colData(myMultiAssay)## DataFrame with 4 rows and 2 columns
## sex age
## <character> <integer>
## Jack M 38
## Jill F 39
## Bob M 40
## Barbara F 41
sampleMap(myMultiAssay)## DataFrame with 16 rows and 3 columns
## assay primary colname
## <factor> <character> <character>
## 1 Affy Jack array1
## 2 Affy Jill array2
## 3 Affy Barbara array3
## 4 Affy Bob array4
## 5 Methyl 450k Jack methyl1
## ... ... ... ...
## 12 Mirna Bob micro3
## 13 CNV gistic Jack mysnparray1
## 14 CNV gistic Jill mysnparray2
## 15 CNV gistic Bob mysnparray3
## 16 CNV gistic Barbara mysnparray4
metadata(myMultiAssay)## list()Note that the ExperimentList class extends the
SimpleList class to add some validity checks specific to
MultiAssayExperiment. It can be used like a list.
Helper function to create a MultiAssayExperiment
object
The prepMultiAssay function helps diagnose common
problems when creating a MultiAssayExperiment object. It
provides error messages and/or warnings in instances where names (either
colnames or ExperimentList element names) are
inconsistent with those found in the sampleMap. Input arguments are the
same as those in the MultiAssayExperiment (i.e.,
ExperimentList, colData,
sampleMap). The resulting output of the
prepMultiAssay function is a list of inputs including a
“metadata$drops” element for names that were not able to be matched.
Instances where ExperimentList is created without names
will prompt an error from prepMultiAssay. Named
ExperimentList elements are essential for checks in
MultiAssayExperiment.
objlist3 <- objlist
(names(objlist3) <- NULL)## NULL
try(prepMultiAssay(objlist3, patient.data, dfmap)$experiments,
outFile = stdout())## Error in prepMultiAssay(objlist3, patient.data, dfmap) :
## ExperimentList does not have names, assign namesNon-matching names may also be present in the
ExperimentList elements and the “assay” column of the
sampleMap. If names only differ by case and are identical
and unique, names will be standardized to lower case and replaced.
## [1] "AFFY" "METHYL 450K" "MIRNA" "CNV GISTIC"
unique(dfmap[, "assay"])## [1] Affy Methyl 450k Mirna CNV gistic
## Levels: Affy Methyl 450k Mirna CNV gistic
prepMultiAssay(objlist3, patient.data, dfmap)$experiments##
## Names in the ExperimentList do not match sampleMap assay
## standardizing will be attempted...## - names set to lowercase## ExperimentList class object of length 4:
## [1] affy: SummarizedExperiment with 2 rows and 4 columns
## [2] methyl 450k: matrix with 2 rows and 5 columns
## [3] mirna: matrix with 4 rows and 3 columns
## [4] cnv gistic: RangedSummarizedExperiment with 5 rows and 4 columnsWhen colnames in the ExperimentList cannot
be matched back to the primary data (colData), these will
be dropped and added to the drops element.
## $`methyl 2k`
## [1] "Jack" "Jill" "Bob" "Bobby"
##
## $`methyl 3k`
## [1] "Jack" "Jane" "Bob"
exampleMap## DataFrame with 5 rows and 3 columns
## assay primary colname
## <factor> <character> <character>
## 1 methyl 2k Jack Jack
## 2 methyl 2k Jill Jill
## 3 methyl 2k Bob Bob
## 4 methyl 3k Jack Jack
## 5 methyl 3k Bob Bob
prepMultiAssay(doubleExp, patient.data, exampleMap)$metadata$drops##
## Not all colnames in the ExperimentList are found in the
## sampleMap, dropping samples from ExperimentList...## $`methyl 2k`
## [1] "Bobby"
##
## $`methyl 3k`
## [1] "Jane"## $`columns.methyl 2k`
## [1] "Bobby"
##
## $`columns.methyl 3k`
## [1] "Jane"A similar operation is performed for checking “primary”
sampleMap names and colData row names. In this
example, we add a row corresponding to “Joe” that does not have a match
in the experimental data.
exMap <- rbind(dfmap,
DataFrame(assay = "New methyl", primary = "Joe",
colname = "Joe"))
invisible(prepMultiAssay(objlist, patient.data, exMap))## Warning in prepMultiAssay(objlist, patient.data, exMap):
## Lengths of names in the ExperimentList and sampleMap
## are not equal##
## Not all names in the primary column of the sampleMap
## could be matched to the colData rownames; see $drops## DataFrame with 1 row and 3 columns
## assay primary colname
## <factor> <character> <character>
## 1 New methyl Joe JoeTo create a MultiAssayExperiment from the results of the
prepMultiAssay function, take each corresponding element
from the resulting list and enter them as arguments to the
MultiAssayExperiment constructor function.
prepped <- prepMultiAssay(objlist, patient.data, exMap)## Warning in prepMultiAssay(objlist, patient.data, exMap):
## Lengths of names in the ExperimentList and sampleMap
## are not equal##
## Not all names in the primary column of the sampleMap
## could be matched to the colData rownames; see $drops## DataFrame with 1 row and 3 columns
## assay primary colname
## <factor> <character> <character>
## 1 New methyl Joe Joe
preppedMulti <- MultiAssayExperiment(prepped$experiments, prepped$colData,
prepped$sampleMap, prepped$metadata)
preppedMulti## A MultiAssayExperiment object of 4 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 2 rows and 4 columns
## [2] Methyl 450k: matrix with 2 rows and 5 columns
## [3] Mirna: matrix with 4 rows and 3 columns
## [4] CNV gistic: RangedSummarizedExperiment with 5 rows and 4 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesAlternatively, use the do.call function to easily create
a MultiAssayExperiment from the output of
prepMultiAssay function:
do.call(MultiAssayExperiment, prepped)## A MultiAssayExperiment object of 4 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 2 rows and 4 columns
## [2] Methyl 450k: matrix with 2 rows and 5 columns
## [3] Mirna: matrix with 4 rows and 3 columns
## [4] CNV gistic: RangedSummarizedExperiment with 5 rows and 4 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesHelper functions to create Bioconductor classes from
raw data
Recent updates to the GenomicRanges and
SummarizedExperiment packages allow the user to create
standard Bioconductor classes from raw data. Raw data read in
as either data.frame or DataFrame can be
converted to GRangesList or
SummarizedExperiment classes depending on the type of
data.
The function to create a GRangesList from a
data.frame, called
makeGRangesListFromDataFrame can be found in the
GenomicRanges package.
makeSummarizedExperimentFromDataFrame is available in the
SummarizedExperiment package. It is also possible to create
a RangedSummarizedExperiment class object from raw data
when ranged data is available.
A simple example can be obtained from the function documentation in
GenomicRanges:
grlls <- list(chr = rep("chr1", nrows), start = seq(11, 15),
end = seq(12, 16), strand = c("+", "-", "+", "*", "*"),
score = seq(1, 5), specimen = c("a", "a", "b", "b", "c"),
gene_symbols = paste0("GENE", letters[seq_len(nrows)]))
grldf <- as.data.frame(grlls, stringsAsFactors = FALSE)
GRL <- makeGRangesListFromDataFrame(grldf, split.field = "specimen",
names.field = "gene_symbols")This can then be converted to a RaggedExperiment object
for a rectangular representation that will conform more easily to the
MultiAssayExperiment API requirements.
RaggedExperiment(GRL)## class: RaggedExperiment
## dim: 5 3
## assays(0):
## rownames(5): GENEa GENEb GENEc GENEd GENEe
## colnames(3): a b c
## colData names(0):Note. See the RaggedExperiment vignette for
more details.
In the SummarizedExperiment package:
sels <- list(chr = rep("chr2", nrows), start = seq(11, 15),
end = seq(12, 16), strand = c("+", "-", "+", "*", "*"),
expr0 = seq(3, 7), expr1 = seq(8, 12), expr2 = seq(12, 16))
sedf <- as.data.frame(sels,
row.names = paste0("GENE", letters[rev(seq_len(nrows))]),
stringsAsFactors = FALSE)
sedf## chr start end strand expr0 expr1 expr2
## GENEe chr2 11 12 + 3 8 12
## GENEd chr2 12 13 - 4 9 13
## GENEc chr2 13 14 + 5 10 14
## GENEb chr2 14 15 * 6 11 15
## GENEa chr2 15 16 * 7 12 16## class: RangedSummarizedExperiment
## dim: 5 3
## metadata(0):
## assays(1): ''
## rownames(5): GENEe GENEd GENEc GENEb GENEa
## rowData names(0):
## colnames(3): expr0 expr1 expr2
## colData names(0):Integrated subsetting across experiments
MultiAssayExperiment allows subsetting by rows, columns,
and assays, rownames, and colnames, across all experiments
simultaneously while guaranteeing continued matching of samples.
Subsetting can be done most compactly by the square bracket method,
or more verbosely and potentially more flexibly by the
subsetBy*() methods.
Subsetting by square bracket [
The three positions within the bracket operator indicate rows, columns, and assays, respectively (pseudocode):
myMultiAssay[rows, columns, assays]For example, to select the gene “ENST00000355076”:
myMultiAssay["ENST00000355076", , ]## A MultiAssayExperiment object of 4 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 1 rows and 4 columns
## [2] Methyl 450k: matrix with 1 rows and 5 columns
## [3] Mirna: matrix with 0 rows and 3 columns
## [4] CNV gistic: RangedSummarizedExperiment with 0 rows and 4 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesThe above operation works across all types of assays, whether
ID-based (e.g. matrix, ExpressionSet,
SummarizedExperiment) or range-based
(e.g. RangedSummarizedExperiment). Note that when using the
bracket method [, the drop argument is TRUE by
default.
You can subset by rows, columns, and assays in a single bracket
operation, and they will be performed in that order (rows, then columns,
then assays). The following selects the ENST00000355076
gene across all samples, then the corresponding samples for the first
two biological specimens in the colData, and finally the
Affy and Methyl 450k assays:
myMultiAssay["ENST00000355076", 1:2, c("Affy", "Methyl 450k")]## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 3 sampleMap rows not in names(experiments)## A MultiAssayExperiment object of 2 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 2:
## [1] Affy: SummarizedExperiment with 1 rows and 2 columns
## [2] Methyl 450k: matrix with 1 rows and 3 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesSubsetting by character, integer, and logical
By columns - character, integer, and logical are all allowed, for example:
myMultiAssay[, "Jack", ]## A MultiAssayExperiment object of 4 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 2 rows and 1 columns
## [2] Methyl 450k: matrix with 2 rows and 2 columns
## [3] Mirna: matrix with 4 rows and 1 columns
## [4] CNV gistic: RangedSummarizedExperiment with 5 rows and 1 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat files
myMultiAssay[, 1, ]## A MultiAssayExperiment object of 4 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 2 rows and 1 columns
## [2] Methyl 450k: matrix with 2 rows and 2 columns
## [3] Mirna: matrix with 4 rows and 1 columns
## [4] CNV gistic: RangedSummarizedExperiment with 5 rows and 1 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat files
myMultiAssay[, c(TRUE, FALSE, FALSE, FALSE), ]## A MultiAssayExperiment object of 4 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 2 rows and 1 columns
## [2] Methyl 450k: matrix with 2 rows and 2 columns
## [3] Mirna: matrix with 4 rows and 1 columns
## [4] CNV gistic: RangedSummarizedExperiment with 5 rows and 1 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesBy assay - character, integer, and logical are allowed:
myMultiAssay[, , "Mirna"]## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 13 sampleMap rows not in names(experiments)
## removing 1 colData rownames not in sampleMap 'primary'## A MultiAssayExperiment object of 1 listed
## experiment with a user-defined name and respective class.
## Containing an ExperimentList class object of length 1:
## [1] Mirna: matrix with 4 rows and 3 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat files
myMultiAssay[, , 3]## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 13 sampleMap rows not in names(experiments)
## removing 1 colData rownames not in sampleMap 'primary'## A MultiAssayExperiment object of 1 listed
## experiment with a user-defined name and respective class.
## Containing an ExperimentList class object of length 1:
## [1] Mirna: matrix with 4 rows and 3 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat files
myMultiAssay[, , c(FALSE, FALSE, TRUE, FALSE, FALSE)]## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 13 sampleMap rows not in names(experiments)
## removing 1 colData rownames not in sampleMap 'primary'## A MultiAssayExperiment object of 1 listed
## experiment with a user-defined name and respective class.
## Containing an ExperimentList class object of length 1:
## [1] Mirna: matrix with 4 rows and 3 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesthe “drop” argument
Specify drop=FALSE to keep assays with zero rows or zero
columns, e.g.:
myMultiAssay["ENST00000355076", , , drop=FALSE]## A MultiAssayExperiment object of 4 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 1 rows and 4 columns
## [2] Methyl 450k: matrix with 1 rows and 5 columns
## [3] Mirna: matrix with 0 rows and 3 columns
## [4] CNV gistic: RangedSummarizedExperiment with 0 rows and 4 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesUsing the default drop=TRUE, assays with no rows or no
columns are removed:
myMultiAssay["ENST00000355076", , , drop=TRUE]## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 7 sampleMap rows not in names(experiments)## A MultiAssayExperiment object of 2 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 2:
## [1] Affy: SummarizedExperiment with 1 rows and 4 columns
## [2] Methyl 450k: matrix with 1 rows and 5 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesMore on subsetting by columns
Experimental samples are stored in the rows of colData
but the columns of elements of ExperimentList, so when we
refer to subsetting by columns, we are referring to columns of the
experimental assays. Subsetting by samples / columns will be more
obvious after recalling the colData:
colData(myMultiAssay)## DataFrame with 4 rows and 2 columns
## sex age
## <character> <integer>
## Jack M 38
## Jill F 39
## Bob M 40
## Barbara F 41Subsetting by samples identifies the selected samples in rows of the
colData DataFrame, then selects all columns of the
ExperimentList corresponding to these rows. Here we use an
integer to keep the first two rows of colData, and all experimental
assays associated to those two primary samples:
myMultiAssay[, 1:2]## A MultiAssayExperiment object of 4 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 2 rows and 2 columns
## [2] Methyl 450k: matrix with 2 rows and 3 columns
## [3] Mirna: matrix with 4 rows and 1 columns
## [4] CNV gistic: RangedSummarizedExperiment with 5 rows and 2 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesNote that the above operation keeps different numbers of columns / samples from each assay, reflecting the reality that some samples may not have been assayed in all experiments, and may have replicates in some.
Columns can be subset using a logical vector. Here the dollar sign
operator ($) accesses one of the columns in
colData.
malesMultiAssay <- myMultiAssay[, myMultiAssay$sex == "M"]
colData(malesMultiAssay)## DataFrame with 2 rows and 2 columns
## sex age
## <character> <integer>
## Jack M 38
## Bob M 40Finally, for special use cases you can exert detailed control of row
or column subsetting, by using a list or
CharacterList to subset. The following creates a
CharacterList of the column names of each assay:
allsamples <- colnames(myMultiAssay)
allsamples## CharacterList of length 4
## [["Affy"]] array1 array2 array3 array4
## [["Methyl 450k"]] methyl1 methyl2 methyl3 methyl4 methyl5
## [["Mirna"]] micro1 micro2 micro3
## [["CNV gistic"]] mysnparray1 mysnparray2 mysnparray3 mysnparray4Now let’s get rid of three Methyl 450k arrays, those in positions 3, 4, and 5:
allsamples[["Methyl 450k"]] <- allsamples[["Methyl 450k"]][-3:-5]
myMultiAssay[, as.list(allsamples), ]## harmonizing input:
## removing 3 sampleMap rows with 'colname' not in colnames of experiments## A MultiAssayExperiment object of 4 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 2 rows and 4 columns
## [2] Methyl 450k: matrix with 2 rows and 2 columns
## [3] Mirna: matrix with 4 rows and 3 columns
## [4] CNV gistic: RangedSummarizedExperiment with 5 rows and 4 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat files
subsetByColumn(myMultiAssay, as.list(allsamples)) #equivalent## harmonizing input:
## removing 3 sampleMap rows with 'colname' not in colnames of experiments## A MultiAssayExperiment object of 4 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 2 rows and 4 columns
## [2] Methyl 450k: matrix with 2 rows and 2 columns
## [3] Mirna: matrix with 4 rows and 3 columns
## [4] CNV gistic: RangedSummarizedExperiment with 5 rows and 4 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesSubsetting assays
You can select certain assays / experiments using subset, by providing a character, logical, or integer vector. An example using character:
myMultiAssay[, , c("Affy", "CNV gistic")]## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 8 sampleMap rows not in names(experiments)## A MultiAssayExperiment object of 2 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 2:
## [1] Affy: SummarizedExperiment with 2 rows and 4 columns
## [2] CNV gistic: RangedSummarizedExperiment with 5 rows and 4 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesYou can subset assays also using logical or integer vectors:
is.cnv <- grepl("CNV", names(experiments(myMultiAssay)))
is.cnv## [1] FALSE FALSE FALSE TRUE
myMultiAssay[, , is.cnv] #logical subsetting## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 12 sampleMap rows not in names(experiments)## A MultiAssayExperiment object of 1 listed
## experiment with a user-defined name and respective class.
## Containing an ExperimentList class object of length 1:
## [1] CNV gistic: RangedSummarizedExperiment with 5 rows and 4 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat files
myMultiAssay[, , which(is.cnv)] #integer subsetting## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 12 sampleMap rows not in names(experiments)## A MultiAssayExperiment object of 1 listed
## experiment with a user-defined name and respective class.
## Containing an ExperimentList class object of length 1:
## [1] CNV gistic: RangedSummarizedExperiment with 5 rows and 4 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesSubsetting rows (features) by IDs, integers, or logicals
Rows of the assays correspond to assay features or measurements, such
as genes. Regardless of whether the assay is ID-based (e.g.,
matrix, ExpressionSet) or range-based (e.g.,
RangedSummarizedExperiment), they can be subset using any
of the following:
a character vector of IDs that will be matched to rownames in each assay
an integer vector that will select rows of this position from each assay. This probably doesn’t make sense unless every
ExperimentListelement represents the same measurements in the same order and will generate an error if any of the integer elements exceeds the number of rows in anyExperimentListelement. The most likely use of integer subsetting would be as aheadfunction, for example to look at the first 6 rows of each assay.a logical vector that will be passed directly to the row subsetting operation for each assay.
a list or List with element names matching those in the
ExperimentList. Each element of the subsetting list will be passed on exactly to subset rows of the corresponding element of theExperimentList.
Any list or List input allows for selective
subsetting. The subsetting is applied only to the matching element names
in the ExperimentList. For example, to only take the first
two rows of the microRNA dataset, we use a named list to
indicate what element we want to subset along with the
drop = FALSE argument.
myMultiAssay[list(Mirna = 1:2), , ]## A MultiAssayExperiment object of 4 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 2 rows and 4 columns
## [2] Methyl 450k: matrix with 2 rows and 5 columns
## [3] Mirna: matrix with 2 rows and 3 columns
## [4] CNV gistic: RangedSummarizedExperiment with 5 rows and 4 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat files
## equivalently
subsetByRow(myMultiAssay, list(Mirna = 1:2))## A MultiAssayExperiment object of 4 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 2 rows and 4 columns
## [2] Methyl 450k: matrix with 2 rows and 5 columns
## [3] Mirna: matrix with 2 rows and 3 columns
## [4] CNV gistic: RangedSummarizedExperiment with 5 rows and 4 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesAgain, these operations always return a
MultiAssayExperiment class, unless drop=TRUE
is passed to the [ backet subset, with any
ExperimentList element not containing the feature having
zero rows.
For example, return a MultiAssayExperiment where Affy
and Methyl 450k contain only “ENST0000035076”” row, and
“Mirna” and “CNV gistic” have zero rows (drop argument is
set to FALSE by default in subsetBy*):
featSub0 <- subsetByRow(myMultiAssay, "ENST00000355076")
featSub1 <- myMultiAssay["ENST00000355076", , drop = FALSE] #equivalent
all.equal(featSub0, featSub1)## [1] TRUE
class(featSub1)## [1] "MultiAssayExperiment"
## attr(,"package")
## [1] "MultiAssayExperiment"
class(experiments(featSub1))## [1] "ExperimentList"
## attr(,"package")
## [1] "MultiAssayExperiment"
experiments(featSub1)## ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 1 rows and 4 columns
## [2] Methyl 450k: matrix with 1 rows and 5 columns
## [3] Mirna: matrix with 0 rows and 3 columns
## [4] CNV gistic: RangedSummarizedExperiment with 0 rows and 4 columnsIn the following, Affy SummarizedExperiment
keeps both rows but with their order reversed, and
Methyl 450k keeps only its second row.
featSubsetted <-
subsetByRow(myMultiAssay, c("ENST00000355076", "ENST00000294241"))
assay(myMultiAssay, 1L)## array1 array2 array3 array4
## ENST00000294241 101 103 105 107
## ENST00000355076 102 104 106 108
assay(featSubsetted, 1L)## array1 array2 array3 array4
## ENST00000355076 102 104 106 108
## ENST00000294241 101 103 105 107Subsetting rows (features) by GenomicRanges
For MultiAssayExperiment objects containing range-based
objects (currently RangedSummarizedExperiment), these can
be subset using a GRanges object, for example:
gr <- GRanges(seqnames = c("chr1", "chr1", "chr2"), strand = c("-", "+", "+"),
ranges = IRanges(start = c(230602, 443625, 934533),
end = c(330701, 443724, 934632)))Now do the subsetting. The function doing the work here is
IRanges::subsetByOverlaps - see its arguments for flexible
types of subsetting by range. The first three arguments here are for
subset, the rest passed on to
IRanges::subsetByOverlaps through “…”:
subsetted <- subsetByRow(myMultiAssay, gr, maxgap = 2L, type = "within")
experiments(subsetted)## ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 0 rows and 4 columns
## [2] Methyl 450k: matrix with 0 rows and 5 columns
## [3] Mirna: matrix with 0 rows and 3 columns
## [4] CNV gistic: RangedSummarizedExperiment with 1 rows and 4 columns
rowRanges(subsetted[[4]])## GRanges object with 1 range and 1 metadata column:
## seqnames ranges strand | feature_id
## <Rle> <IRanges> <Rle> | <character>
## a chr1 317182-317281 - | ID\\001
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengthsSquare bracket subsetting can still be used here, but passing on
arguments to IRanges::subsetByOverlaps through “…” is
simpler using subsetByRow().
Subsetting is endomorphic
subsetByRow, subsetByColumn,
subsetByAssay, and square bracket subsetting are all
“endomorphic” operations, in that they always return another
MultiAssayExperiment object.
Double-bracket subsetting to select experiments
A double-bracket subset operation refers to an experiment, and will
return the object contained within an ExperimentList
element. It is not endomorphic. For example, the first
ExperimentList element is called “Affy” and contains a
SummarizedExperiment:
names(myMultiAssay)## [1] "Affy" "Methyl 450k" "Mirna" "CNV gistic"
myMultiAssay[[1]]## class: SummarizedExperiment
## dim: 2 4
## metadata(0):
## assays(1): ''
## rownames(2): ENST00000294241 ENST00000355076
## rowData names(0):
## colnames(4): array1 array2 array3 array4
## colData names(1): slope53
myMultiAssay[["Affy"]]## class: SummarizedExperiment
## dim: 2 4
## metadata(0):
## assays(1): ''
## rownames(2): ENST00000294241 ENST00000355076
## rowData names(0):
## colnames(4): array1 array2 array3 array4
## colData names(1): slope53Helpers for data clean-up and management
complete.cases
The complete.cases function returns a logical vector of
colData rows identifying which primary units have data for
all experiments. Recall that myMultiAssay provides data for
four individuals:
colData(myMultiAssay)## DataFrame with 4 rows and 2 columns
## sex age
## <character> <integer>
## Jack M 38
## Jill F 39
## Bob M 40
## Barbara F 41Of these, only Jack has data for all 5 experiments:
complete.cases(myMultiAssay)## [1] TRUE FALSE TRUE TRUEBut all four have complete cases for Affy and Methyl 450k:
complete.cases(myMultiAssay[, , 1:2])## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 7 sampleMap rows not in names(experiments)## [1] TRUE TRUE TRUE TRUEThis output can be used to select individuals with complete data:
myMultiAssay[, complete.cases(myMultiAssay), ]## A MultiAssayExperiment object of 4 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 2 rows and 3 columns
## [2] Methyl 450k: matrix with 2 rows and 4 columns
## [3] Mirna: matrix with 4 rows and 3 columns
## [4] CNV gistic: RangedSummarizedExperiment with 5 rows and 3 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat files
replicated
The replicated function identifies primary
column values or biological units that have multiple observations per
assay. It returns a list of
LogicalLists that indicate what biological units have one
or more replicate measurements. This output is used for merging
replicates by default.
replicated(myMultiAssay)## $Affy
## LogicalList of length 4
## [["Barbara"]] FALSE FALSE FALSE FALSE
## [["Bob"]] FALSE FALSE FALSE FALSE
## [["Jack"]] FALSE FALSE FALSE FALSE
## [["Jill"]] FALSE FALSE FALSE FALSE
##
## $`Methyl 450k`
## LogicalList of length 4
## [["Barbara"]] FALSE FALSE FALSE FALSE FALSE
## [["Bob"]] FALSE FALSE FALSE FALSE FALSE
## [["Jack"]] TRUE TRUE FALSE FALSE FALSE
## [["Jill"]] FALSE FALSE FALSE FALSE FALSE
##
## $Mirna
## LogicalList of length 3
## [["Barbara"]] FALSE FALSE FALSE
## [["Bob"]] FALSE FALSE FALSE
## [["Jack"]] FALSE FALSE FALSE
##
## $`CNV gistic`
## LogicalList of length 4
## [["Barbara"]] FALSE FALSE FALSE FALSE
## [["Bob"]] FALSE FALSE FALSE FALSE
## [["Jack"]] FALSE FALSE FALSE FALSE
## [["Jill"]] FALSE FALSE FALSE FALSE
intersectRows
The intersectRows function takes all common rownames
across all experiments and returns a MultiAssayExperiment
with those rows.
(ensmblMatches <- intersectRows(myMultiAssay[, , 1:2]))## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 7 sampleMap rows not in names(experiments)## A MultiAssayExperiment object of 2 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 2:
## [1] Affy: SummarizedExperiment with 1 rows and 4 columns
## [2] Methyl 450k: matrix with 1 rows and 5 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat files
rownames(ensmblMatches)## CharacterList of length 2
## [["Affy"]] ENST00000355076
## [["Methyl 450k"]] ENST00000355076
intersectColumns
A call to intersectColumns returns another
MultiAssayExperiment where the columns of each element of
the ExperimentList correspond exactly to the rows of
colData. In many cases, this operation returns a 1-to-1
correspondence of samples to patients for each experiment assay unless
replicates are present in the data.
intersectColumns(myMultiAssay)## A MultiAssayExperiment object of 4 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 2 rows and 3 columns
## [2] Methyl 450k: matrix with 2 rows and 4 columns
## [3] Mirna: matrix with 4 rows and 3 columns
## [4] CNV gistic: RangedSummarizedExperiment with 5 rows and 3 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat files
mergeReplicates
The mergeReplicates function allows the user to specify
a function (default: mean) for combining replicate columns
in each assay element. This can be combined with
intersectColumns to create a
MultiAssayExperiment object with one measurement in each
experiment per biological unit.
mergeReplicates(intersectColumns(myMultiAssay))## harmonizing input:
## removing 1 sampleMap rows with 'colname' not in colnames of experiments## A MultiAssayExperiment object of 4 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 4:
## [1] Affy: SummarizedExperiment with 2 rows and 3 columns
## [2] Methyl 450k: matrix with 2 rows and 3 columns
## [3] Mirna: matrix with 4 rows and 3 columns
## [4] CNV gistic: RangedSummarizedExperiment with 5 rows and 3 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filescombine c
The combine c function allows the user to append an
experiment to the list of experiments already present in
MultiAssayExperiment. In the case that additional
observations on the same set of samples were performed, the
c function can conveniently be referenced to an existing
assay that contains the same ordering of sample measurements.
The mapFrom argument indicates what experiment has the
exact same organization of samples that will be introduced by the new
experiment dataset. If the number of columns in the new experiment do
not match those in the reference experiment, an error will be
thrown.
Here we introduce a toy dataset created on the fly:
c(myMultiAssay, ExpScores = matrix(1:8, ncol = 4,
dim = list(c("ENSMBL0001", "ENSMBL0002"), paste0("pt", 1:4))),
mapFrom = 1L)## Warning: Assuming column order in the data provided
## matches the order in 'mapFrom' experiment(s) colnames## A MultiAssayExperiment object of 5 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 5:
## [1] Affy: SummarizedExperiment with 2 rows and 4 columns
## [2] Methyl 450k: matrix with 2 rows and 5 columns
## [3] Mirna: matrix with 4 rows and 3 columns
## [4] CNV gistic: RangedSummarizedExperiment with 5 rows and 4 columns
## [5] ExpScores: matrix with 2 rows and 4 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesNote: Alternatively, a sampleMap for the
additional dataset can be provided.
Extractor functions
Extractor functions convert a MultiAssayExperiment into
other forms that are convenient for analyzing. These would normally be
called after any desired subsetting has been performed.
getWithColData
Provides a single assay along with any associated ‘colData’ columns while keeping the assay class constant.
(affex <- getWithColData(myMultiAssay, 1L))## Warning: 'experiments' dropped; see 'drops()'## class: SummarizedExperiment
## dim: 2 4
## metadata(0):
## assays(1): ''
## rownames(2): ENST00000294241 ENST00000355076
## rowData names(0):
## colnames(4): array1 array2 array3 array4
## colData names(3): slope53 sex age
colData(affex)## DataFrame with 4 rows and 3 columns
## slope53 sex age
## <numeric> <character> <integer>
## array1 0.106685 M 38
## array2 -0.587014 F 39
## array3 -0.327854 F 41
## array4 -0.085361 M 40
class(affex)## [1] "SummarizedExperiment"
## attr(,"package")
## [1] "SummarizedExperiment"It will error when the target data class does not support a
colData replacement method, meaning that it typically works
with SummarizedExperiment and RaggedExperiment
assays and their extensions.
longForm & wideFormat
Produces long (default) or wide
DataFrame objects. The following produces a long
DataFrame (the default) for the first two assays:
longForm(myMultiAssay[, , 1:2])## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 7 sampleMap rows not in names(experiments)## DataFrame with 18 rows and 5 columns
## assay primary rowname colname value
## <character> <character> <character> <factor> <integer>
## 1 Affy Jack ENST00000294241 array1 101
## 2 Affy Jack ENST00000355076 array1 102
## 3 Affy Jill ENST00000294241 array2 103
## 4 Affy Jill ENST00000355076 array2 104
## 5 Affy Barbara ENST00000294241 array3 105
## ... ... ... ... ... ...
## 14 Methyl 450k Jill ENST00000383706 methyl3 6
## 15 Methyl 450k Barbara ENST00000355076 methyl4 7
## 16 Methyl 450k Barbara ENST00000383706 methyl4 8
## 17 Methyl 450k Bob ENST00000355076 methyl5 9
## 18 Methyl 450k Bob ENST00000383706 methyl5 10This is especially useful for performing regression against patient
or sample data from colData using the
pDataCols argument:
longForm(myMultiAssay[, , 1:2], colDataCols="age")## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 7 sampleMap rows not in names(experiments)## DataFrame with 18 rows and 6 columns
## assay primary rowname colname value age
## <character> <character> <character> <factor> <integer> <integer>
## 1 Affy Jack ENST00000294241 array1 101 38
## 2 Affy Jack ENST00000355076 array1 102 38
## 3 Affy Jill ENST00000294241 array2 103 39
## 4 Affy Jill ENST00000355076 array2 104 39
## 5 Affy Barbara ENST00000294241 array3 105 41
## ... ... ... ... ... ... ...
## 14 Methyl 450k Jill ENST00000383706 methyl3 6 39
## 15 Methyl 450k Barbara ENST00000355076 methyl4 7 41
## 16 Methyl 450k Barbara ENST00000383706 methyl4 8 41
## 17 Methyl 450k Bob ENST00000355076 methyl5 9 40
## 18 Methyl 450k Bob ENST00000383706 methyl5 10 40The “wide” format is useful for calculating correlations or
performing regression against different genomic features. Wide format is
in general not possible with replicate measurements, so we demonstrate
on the cleaned MultiAssayExperiment for the first 5
columns:
maemerge <- mergeReplicates(intersectColumns(myMultiAssay))## harmonizing input:
## removing 1 sampleMap rows with 'colname' not in colnames of experiments
wideFormat(maemerge, colDataCols="sex")[, 1:5]## DataFrame with 3 rows and 5 columns
## primary sex Affy_ENST00000294241 Affy_ENST00000355076
## <character> <character> <numeric> <numeric>
## 1 Jack M 101 102
## 2 Bob M 107 108
## 3 Barbara F 105 106
## CNV.gistic_a
## <numeric>
## 1 201.632
## 2 9200.135
## 3 9970.694
assay / assays
The assay (singular) function takes a particular
experiment and returns a matrix. By default, it will return the
first experiment as a matrix.
assay(myMultiAssay)## array1 array2 array3 array4
## ENST00000294241 101 103 105 107
## ENST00000355076 102 104 106 108The assays (plural) function returns a
SimpleList of data matrices from the
ExperimentList:
assays(myMultiAssay)## List of length 4
## names(4): Affy Methyl 450k Mirna CNV gisticThe Cancer Genome Atlas and MultiAssayExperiment
Our most recent efforts include the release of the experiment data
package, curatedTCGAData. This package will allow users to
selectively download cancer datasets from The Cancer Genome Atlas (TCGA)
and represent the data as MultiAssayExperiment objects.
Please see the package vignette for more details.
BiocManager::install("curatedTCGAData")Dimension names: rownames and
colnames
rownames and colnames return a
CharacterList of row names and column names across all the
assays. A CharacterList is an efficient alternative to
list used when each element contains a character vector. It
also provides a nice show method:
rownames(myMultiAssay)## CharacterList of length 4
## [["Affy"]] ENST00000294241 ENST00000355076
## [["Methyl 450k"]] ENST00000355076 ENST00000383706
## [["Mirna"]] hsa-miR-21 hsa-miR-191 hsa-miR-148a hsa-miR148b
## [["CNV gistic"]] a b c d e
colnames(myMultiAssay)## CharacterList of length 4
## [["Affy"]] array1 array2 array3 array4
## [["Methyl 450k"]] methyl1 methyl2 methyl3 methyl4 methyl5
## [["Mirna"]] micro1 micro2 micro3
## [["CNV gistic"]] mysnparray1 mysnparray2 mysnparray3 mysnparray4Requirements for support of additional data classes
Any data classes in the ExperimentList object must
support the following methods:
dimnames[dim()
Here is what happens if one of the methods does not:
objlist2 <- objlist
objlist2[[2]] <- as.vector(objlist2[[2]])
tryCatch(
MultiAssayExperiment(objlist2, patient.data, dfmap),
error = function(e) {
conditionMessage(e)
}
)## [1] "invalid class \"ExperimentList\" object: \n 'integer' class is not supported, use a rectangular class"Application Programming Interface (API)
For more information on the formal API of
MultiAssayExperiment, please see the API
wiki document on GitHub. An API package is available for download on
GitHub via install("waldronlab/MultiAssayShiny"). It
provides visual exploration of available methods in
MultiAssayExperiment.
Methods for MultiAssayExperiment
The following methods are defined for
MultiAssayExperiment:
methods(class="MultiAssayExperiment")## [1] [ [[ [[<- [<-
## [5] $ $<- anyReplicated assay
## [9] assays c coerce colData
## [13] colData<- colnames<- complete.cases dimnames
## [17] drops drops<- experiments experiments<-
## [21] exportClass hasRowData hasRowRanges intersectByRowData
## [25] isEmpty length longForm longFormat
## [29] mergeReplicates metadata metadata<- names
## [33] names<- replicated replicates sampleMap
## [37] sampleMap<- show showReplicated splitAssays
## [41] subsetByAssay subsetByColData subsetByColumn subsetByRow
## [45] subsetByRowData updateObject
## see '?methods' for accessing help and source codesessionInfo()
## R version 4.5.2 (2025-10-31)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.3 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: Etc/UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] RaggedExperiment_1.34.0 MultiAssayExperiment_1.36.2
## [3] SummarizedExperiment_1.40.0 Biobase_2.70.0
## [5] GenomicRanges_1.62.1 Seqinfo_1.0.0
## [7] IRanges_2.44.0 S4Vectors_0.48.0
## [9] BiocGenerics_0.56.0 generics_0.1.4
## [11] MatrixGenerics_1.22.0 matrixStats_1.5.0
## [13] BiocStyle_2.38.0
##
## loaded via a namespace (and not attached):
## [1] tidyr_1.3.2 sass_0.4.10 SparseArray_1.10.9
## [4] stringi_1.8.7 lattice_0.22-9 magrittr_2.0.4
## [7] digest_0.6.39 evaluate_1.0.5 grid_4.5.2
## [10] bookdown_0.46 fastmap_1.2.0 plyr_1.8.9
## [13] jsonlite_2.0.0 Matrix_1.7-5 BiocManager_1.30.27
## [16] purrr_1.2.1 textshaping_1.0.5 jquerylib_0.1.4
## [19] abind_1.4-8 cli_3.6.5 rlang_1.1.7
## [22] XVector_0.50.0 withr_3.0.2 cachem_1.1.0
## [25] DelayedArray_0.36.0 yaml_2.3.12 otel_0.2.0
## [28] BiocBaseUtils_1.12.0 S4Arrays_1.10.1 tools_4.5.2
## [31] reshape2_1.4.5 dplyr_1.2.0 vctrs_0.7.2
## [34] R6_2.6.1 lifecycle_1.0.5 stringr_1.6.0
## [37] fs_2.0.1 htmlwidgets_1.6.4 ragg_1.5.2
## [40] pkgconfig_2.0.3 desc_1.4.3 pillar_1.11.1
## [43] pkgdown_2.2.0 bslib_0.10.0 Rcpp_1.1.1
## [46] glue_1.8.0 systemfonts_1.3.2 tidyselect_1.2.1
## [49] tibble_3.3.1 xfun_0.57 knitr_1.51
## [52] htmltools_0.5.9 rmarkdown_2.31 compiler_4.5.2