HMP16SData
Lucas Schiffer
Graduate School of Public Health and Health Policy, City University of New York, New York, NYInstitute for Implementation Science in Population Health, City University of New York, New York, NYRimsha Azhar
Graduate School of Public Health and Health Policy, City University of New York, New York, NYInstitute for Implementation Science in Population Health, City University of New York, New York, NYMarcel Ramos
Graduate School of Public Health and Health Policy, City University of New York, New York, NYInstitute for Implementation Science in Population Health, City University of New York, New York, NYRoswell Park Cancer Institute, University of Buffalo, Buffalo, NYLudwig Geistlinger
Graduate School of Public Health and Health Policy, City University of New York, New York, NYInstitute for Implementation Science in Population Health, City University of New York, New York, NYLevi Waldron
Graduate School of Public Health and Health Policy, City University of New York, New York, NYInstitute for Implementation Science in Population Health, City University of New York, New York, NYMarch 26, 2026
Source:vignettes/HMP16SData.Rmd
HMP16SData.RmdAbstract
HMP16SData is a Bioconductor ExperimentData package of the Human Microbiome Project (HMP) 16S rRNA sequencing data for variable regions 1–3 and 3–5. Raw data files are provided in the package as downloaded from the HMP Data Analysis and Coordination Center. Processed data is provided as SummarizedExperiment class objects via ExperimentHub.
Publications
Schiffer, L. et al. HMP16SData: Efficient Access to the Human Microbiome Project through Bioconductor. Am. J. Epidemiol. (2019).
Griffith, J. C. & Morgan, X. C. Invited Commentary: Improving accessibility of the Human Microbiome Project data through integration with R/Bioconductor. Am. J. Epidemiol. (2019).
Waldron, L. et al. Waldron et al. Reply to “Commentary on the HMP16SData Bioconductor Package”. Am. J. Epidemiol. (2019).
Prerequisites
The following knitr options will be used in this vignette to provide the most useful and concise output.
knitr::opts_chunk$set(message = FALSE)The following packages will be used in this vignette to provide demonstrative examples of what a user might do with HMP16SData.
library(HMP16SData)
library(phyloseq)
library(magrittr)
library(ggplot2)
library(tibble)
library(dplyr)
library(dendextend)
library(circlize)
library(ExperimentHub)
library(gridExtra)
library(cowplot)
library(readr)
library(haven)Pipe operators from the magrittr package are used in this vignette to provide the most elegant and concise syntax. See the magrittr vignette if the syntax is unclear.
Introduction
HMP16SData
is a Bioconductor ExperimentData package of the Human Microbiome Project
(HMP) 16S rRNA sequencing data for variable regions 1–3 and 3–5. Raw
data files are provided in the package as downloaded from the HMP Data Analysis and Coordination
Center. Processed data is provided as
SummarizedExperiment class objects via ExperimentHub.
HMP16SData can be installed using BiocManager as follows.
BiocManager::install("HMP16SData")Once installed, HMP16SData
provides two functions to access data – one for variable region 1–3 and
another for variable region 3–5. When called, as follows, the functions
will download data from an ExperimentHub
Amazon S3 (Simple Storage Service) bucket over https or
load data from a local cache.
V13()## class: SummarizedExperiment
## dim: 43140 2898
## metadata(2): experimentData phylogeneticTree
## assays(1): 16SrRNA
## rownames(43140): OTU_97.1 OTU_97.10 ... OTU_97.9997 OTU_97.9999
## rowData names(7): CONSENSUS_LINEAGE SUPERKINGDOM ... FAMILY GENUS
## colnames(2898): 700013549 700014386 ... 700114963 700114965
## colData names(7): RSID VISITNO ... HMP_BODY_SUBSITE SRS_SAMPLE_ID
V35()## class: SummarizedExperiment
## dim: 45383 4743
## metadata(2): experimentData phylogeneticTree
## assays(1): 16SrRNA
## rownames(45383): OTU_97.1 OTU_97.10 ... OTU_97.9998 OTU_97.9999
## rowData names(7): CONSENSUS_LINEAGE SUPERKINGDOM ... FAMILY GENUS
## colnames(4743): 700013549 700014386 ... 700114717 700114750
## colData names(7): RSID VISITNO ... HMP_BODY_SUBSITE SRS_SAMPLE_IDThe two data sets are represented as
SummarizedExperiment objects, a standard Bioconductor class
that is amenable to subsetting and analysis. To maintain brevity,
details of the SummarizedExperiment class are not outlined
here but the SummarizedExperiment
package provides an excellent vignette.
Features
Frequency Table Generation
Sometimes it is desirable to provide a quick summary of key
demographic variables and to make the process easier HMP16SData
provides a function, table_one, to do so. It returns a
data.frame or a list of
data.frame objects that have been transformed to make a
publication ready table.
## Visit Number Sex Run Center
## 700013549 One Female Baylor College of Medicine
## 700014386 One Male Baylor College of Medicine, Broad Institute
## 700014403 One Male Baylor College of Medicine, Broad Institute
## 700014409 One Male Baylor College of Medicine, Broad Institute
## 700014412 One Male Baylor College of Medicine, Broad Institute
## 700014415 One Male Baylor College of Medicine, Broad Institute
## HMP Body Site HMP Body Subsite
## 700013549 Gastrointestinal Tract Stool
## 700014386 Gastrointestinal Tract Stool
## 700014403 Oral Saliva
## 700014409 Oral Tongue Dorsum
## 700014412 Oral Hard Palate
## 700014415 Oral Buccal MucosaIf a list is passed to table_one, its
elements must be named so that the named elements can be used by the
kable_one function. The kable_one function
will produce an HTML table for vignettes such as the one
shown below.
| N | % | N | % | |
|---|---|---|---|---|
| Visit Number | ||||
| One | 1,642 | 56.66 | 2,822 | 59.50 |
| Two | 1,244 | 42.93 | 1,897 | 40.00 |
| Three | 12 | 0.41 | 24 | 0.51 |
| Sex | ||||
| Female | 1,521 | 52.48 | 2,188 | 46.13 |
| Male | 1,377 | 47.52 | 2,555 | 53.87 |
| Run Center | ||||
| Genome Sequencing Center at Washington University | 1,717 | 59.25 | 1,539 | 32.45 |
| J. Craig Venter Institute | 506 | 17.46 | 1,009 | 21.27 |
| Broad Institute | 0 | 0.00 | 1,078 | 22.73 |
| Baylor College of Medicine | 289 | 9.97 | 696 | 14.67 |
| J. Craig Venter Institute, Genome Sequencing Center at Washington University | 97 | 3.35 | 93 | 1.96 |
| Baylor College of Medicine, Genome Sequencing Center at Washington University | 91 | 3.14 | 90 | 1.90 |
| Baylor College of Medicine, Broad Institute | 76 | 2.62 | 103 | 2.17 |
| J. Craig Venter Institute, Broad Institute | 86 | 2.97 | 75 | 1.58 |
| Baylor College of Medicine, J. Craig Venter Institute | 15 | 0.52 | 40 | 0.84 |
| Broad Institute, Baylor College of Medicine | 13 | 0.45 | 13 | 0.27 |
| Genome Sequencing Center at Washington University, Baylor College of Medicine | 6 | 0.21 | 6 | 0.13 |
| Genome Sequencing Center at Washington University, J. Craig Venter Institute | 1 | 0.03 | 1 | 0.02 |
| Missing | 1 | 0.03 | 0 | 0.00 |
| HMP Body Site | ||||
| Oral | 1,622 | 55.97 | 2,774 | 58.49 |
| Skin | 664 | 22.91 | 990 | 20.87 |
| Urogenital Tract | 264 | 9.11 | 391 | 8.24 |
| Gastrointestinal Tract | 187 | 6.45 | 319 | 6.73 |
| Airways | 161 | 5.56 | 269 | 5.67 |
| HMP Body Subsite | ||||
| Tongue Dorsum | 190 | 6.56 | 316 | 6.66 |
| Stool | 187 | 6.45 | 319 | 6.73 |
| Supragingival Plaque | 189 | 6.52 | 313 | 6.60 |
| Right Retroauricular Crease | 187 | 6.45 | 297 | 6.26 |
| Attached Keratinized Gingiva | 181 | 6.25 | 313 | 6.60 |
| Left Retroauricular Crease | 186 | 6.42 | 285 | 6.01 |
| Buccal Mucosa | 183 | 6.31 | 312 | 6.58 |
| Palatine Tonsils | 186 | 6.42 | 312 | 6.58 |
| Subgingival Plaque | 183 | 6.31 | 309 | 6.51 |
| Throat | 170 | 5.87 | 307 | 6.47 |
| Hard Palate | 178 | 6.14 | 302 | 6.37 |
| Saliva | 162 | 5.59 | 290 | 6.11 |
| Anterior Nares | 161 | 5.56 | 269 | 5.67 |
| Right Antecubital Fossa | 146 | 5.04 | 207 | 4.36 |
| Left Antecubital Fossa | 145 | 5.00 | 201 | 4.24 |
| Mid Vagina | 89 | 3.07 | 133 | 2.80 |
| Posterior Fornix | 88 | 3.04 | 133 | 2.80 |
| Vaginal Introitus | 87 | 3.00 | 125 | 2.64 |
Straightforward Subsetting
The SummarizedExperiment container provides for
straightforward subsetting by data or metadata variables using either
the subset function or [ methods – see the
SummarizedExperiment
vignette for additional details. Shown below, the variable region 3–5
data set is subset to include only stool samples.
## class: SummarizedExperiment
## dim: 45383 319
## metadata(2): experimentData phylogeneticTree
## assays(1): 16SrRNA
## rownames(45383): OTU_97.1 OTU_97.10 ... OTU_97.9998 OTU_97.9999
## rowData names(7): CONSENSUS_LINEAGE SUPERKINGDOM ... FAMILY GENUS
## colnames(319): 700013549 700014386 ... 700114717 700114750
## colData names(7): RSID VISITNO ... HMP_BODY_SUBSITE SRS_SAMPLE_IDHMP Controlled-Access Participant Data
Most participant data from the HMP study is controlled through the
National Center for Biotechnology Information (NCBI) database of
Genotypes and Phenotypes (dbGaP). HMP16SData
provides a data dictionary translated from dbGaP XML files
for the seven different controlled-access data tables related to the
HMP. See ?HMP16SData::dictionary for details of these
source data tables, and View(dictionary) to view the
complete data dictionary. Several steps are required to access the data
tables, but the process is greatly simplified by HMP16SData.
Apply for dbGaP Access
You must make a controlled-access application through https://dbgap.ncbi.nlm.nih.gov for:
HMP Core Microbiome Sampling Protocol A (HMP-A) (phs000228.v4.p1)
Once approved, browse to https://dbgap.ncbi.nlm.nih.gov, sign in, and select the
option “get dbGaP repository key” to download your
*.ngc repository key. This is all you need from the dbGaP
website.
Install the SRA Toolkit
You must also install the NCBI SRA Toolkit, which will be used in the background for downloading and decrypting controlled-access data.
There are shortcuts for common platforms:
- Debian/Ubuntu:
apt install sra-toolkit - macOS:
brew install sratoolkit
For macOS, the brew command does not come installed by
default and requires installation of the homebrew package manager.
Instructions are available at https://tinyurl.com/ybeqwl8f.
For Windows, binary installation is necessary and instructions are available at https://tinyurl.com/y845ppaa.
Merge with HMP16SData
The attach_dbGap() function takes a HMP16SData
SummarizedExperiment object as its first argument and the
path to a dbGaP repository key as its second argument. It performs
download, decryption, and merging of all available controlled-access
participant data with a single function call.
V35_stool_protected <-
V35_stool %>%
attach_dbGaP("~/prj_12146.ngc")The returned V35_stool_protected object contains
controlled-access participant data as additional columns in its
colData slot.
colData(V35_stool_protected)Analysis Using the phyloseq Package
The phyloseq package provides an extensive suite of methods to analyze microbiome data.
For those familiar with both the HMP and phyloseq,
you may recall that an alternative phyloseq class object
containing the HMP variable region 3–5 data has been made available by
Joey McMurdie at https://joey711.github.io/phyloseq-demo/HMPv35.RData.
However, this object is not compatible with the methods documented here
for integration with dbGaP controlled-access participant data, shotgun
metagenomic data, or variable region 1–3 data. For that reason, we would
encourage the use of the HMP16SData
SummarizedExperiment class objects with the phyloseq
package.
To demonstrate how HMP16SData
could be used as a control or comparison cohort in microbime data
analyses, we will demonstrate basic comparisons of the palatine tonsils
and stool body subsites using the phyloseq
package. We first create and subset two
SummarizedExperiment objects from HMP16SData
to include only the relevant body subsites.
V13_tonsils <-
V13() %>%
subset(select = HMP_BODY_SUBSITE == "Palatine Tonsils")
V13_stool <-
V13() %>%
subset(select = HMP_BODY_SUBSITE == "Stool")While these objects are both from the HMP16SData package, a user would potentially be comparing to their own data and only need a single object from the package.
Coercion to phyloseq Objects
The SummarizedExperiment class objects can then be
coerced to phyloseq class objects containing count data,
sample (participant) data, taxonomy, and phylogenetic trees using the
as_phyloseq function.
V13_tonsils_phyloseq <-
as_phyloseq(V13_tonsils)
V13_stool_phyloseq <-
as_phyloseq(V13_stool)The analysis of all the samples in these two phyloseq
objects would be rather computationally intensive. So to preform the
analysis in a more timely manner, a function,
sample_samples, is written here to take a sample of the
samples available in each phyloseq object.
sample_samples <- function(x, size) {
sampled_names <-
sample_names(x) %>%
sample(size)
prune_samples(sampled_names, x)
}Each phyloseq object is then sampled to contain only
twenty-five samples.
A “Study” identifier is then added to the sample_data of
each phyloseq object to be used for stratification in
analysis. In the case that a user were comparing the HMP samples to
their own data, an identifier would be added in the same manner.
sample_data(V13_tonsils_phyloseq)$Study <- "Tonsils"
sample_data(V13_stool_phyloseq)$Study <- "Stool"Once the two phyloseq objects have been sampled and
their sample_data has been augmented, they can be merged
into a single phyloseq object using the
merge_phyloseq command.
V13_phyloseq <-
merge_phyloseq(V13_tonsils_phyloseq, V13_stool_phyloseq)Finally, because the V13 data were subset and sampled, taxa with no
relative abundance are present in the merged object. These are removed
using the prune_taxa command to avoid warnings during
analysis.
V13_phyloseq %<>%
taxa_sums() %>%
is_greater_than(0) %>%
prune_taxa(V13_phyloseq)The resulting V13_phyloseq object can then be analyzed
quickly and easily.
Alpha Diversity Analysis
Alpha diversity measures the taxonomic variation within a sample and
phyloseq
provides a method, plot_richness, to plot various alpha
diversity measures.
First a vector of richness (i.e. alpha diversity) measures is created
to be passed to the plot_richness method.
richness_measures <-
c("Observed", "Shannon", "Simpson")The V13_phyloseq object and the
richness_measures vector are then passed to the
plot_richness method to construct a box plot of the three
alpha diversity measures. Additional ggplot2
syntax is used to control the presentational aspects of the plot.
V13_phyloseq %>%
plot_richness(x = "Study", color = "Study", measures = richness_measures) +
stat_boxplot(geom ="errorbar") +
geom_boxplot() +
theme_bw() +
theme(axis.title.x = element_blank(), legend.position = "none")## Warning: `aes_string()` was deprecated in ggplot2 3.0.0.
## ℹ Please use tidy evaluation idioms with `aes()`.
## ℹ See also `vignette("ggplot2-in-packages")` for more information.
## ℹ The deprecated feature was likely used in the phyloseq package.
## Please report the issue at <https://github.com/joey711/phyloseq/issues>.
## This warning is displayed once per session.
## Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
## generated.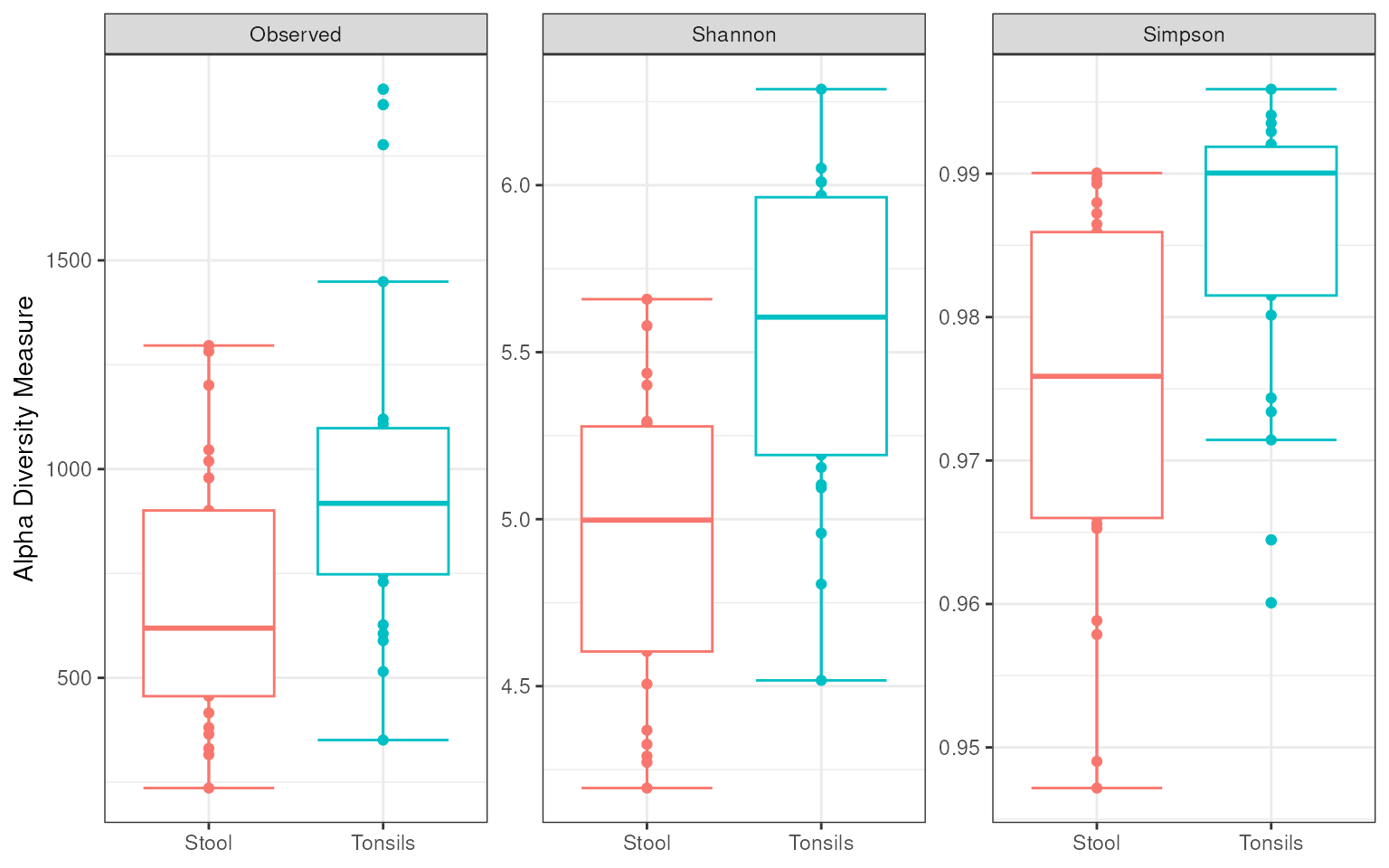
Beta Diversity Analysis
Beta diversity measures the taxonomic variation between samples by
calculating the dissimilarity of clade relative abundances. The phyloseq
package provides a method, distance, to calculate various
dissimilarity measures, such as Bray–Curtis dissimilarity. Once
dissimilarity has been calculated, samples can then be clustered and
represented as a dendrogram.
V13_dendrogram <-
distance(V13_phyloseq, method = "bray") %>%
hclust() %>%
as.dendrogram()However, coercion to a dendrogram object results in the
lost of sample_data present in the phyloseq
object which is needed for plotting. A data.frame of this
sample_data can be extracted from the phyloseq
object as follows.
V13_sample_data <-
sample_data(V13_phyloseq) %>%
data.frame()Samples in the the plots will be identified by “PSN”” (Primary Sample
Number) and “Study”. So, additional columns to denote the colors and
shapes of leaves and labels are added to the data.frame
using dplyr
syntax.
V13_sample_data %<>%
rownames_to_column(var = "PSN") %>%
mutate(labels_col = if_else(Study == "Stool", "#F8766D", "#00BFC4")) %>%
mutate(leaves_col = if_else(Study == "Stool", "#F8766D", "#00BFC4")) %>%
mutate(leaves_pch = if_else(Study == "Stool", 16, 17))Additionally, the order of samples in the dendrogram and
data.frame objects is different and a vector to sort
samples is constructed as follows.
The label and leaf color and shape columns of the
data.frame object can then be coerced to vectors and sorted
according to the sample order of the dendrogram object.
labels_col <- V13_sample_data$labels_col[V13_sample_order]
leaves_col <- V13_sample_data$leaves_col[V13_sample_order]
leaves_pch <- V13_sample_data$leaves_pch[V13_sample_order]The dendextend
package is then used to add these vectors to the dendrogram
object as metadata which will be used for plotting.
V13_dendrogram %<>%
set("labels_col", labels_col) %>%
set("leaves_col", leaves_col) %>%
set("leaves_pch", leaves_pch)Finally, the dendextend
package provides a method, circlize_dendrogram, to produce
a circular dendrogram, using the circlize
package, with a single line of code.
V13_dendrogram %>%
circlize_dendrogram(labels_track_height = 0.2)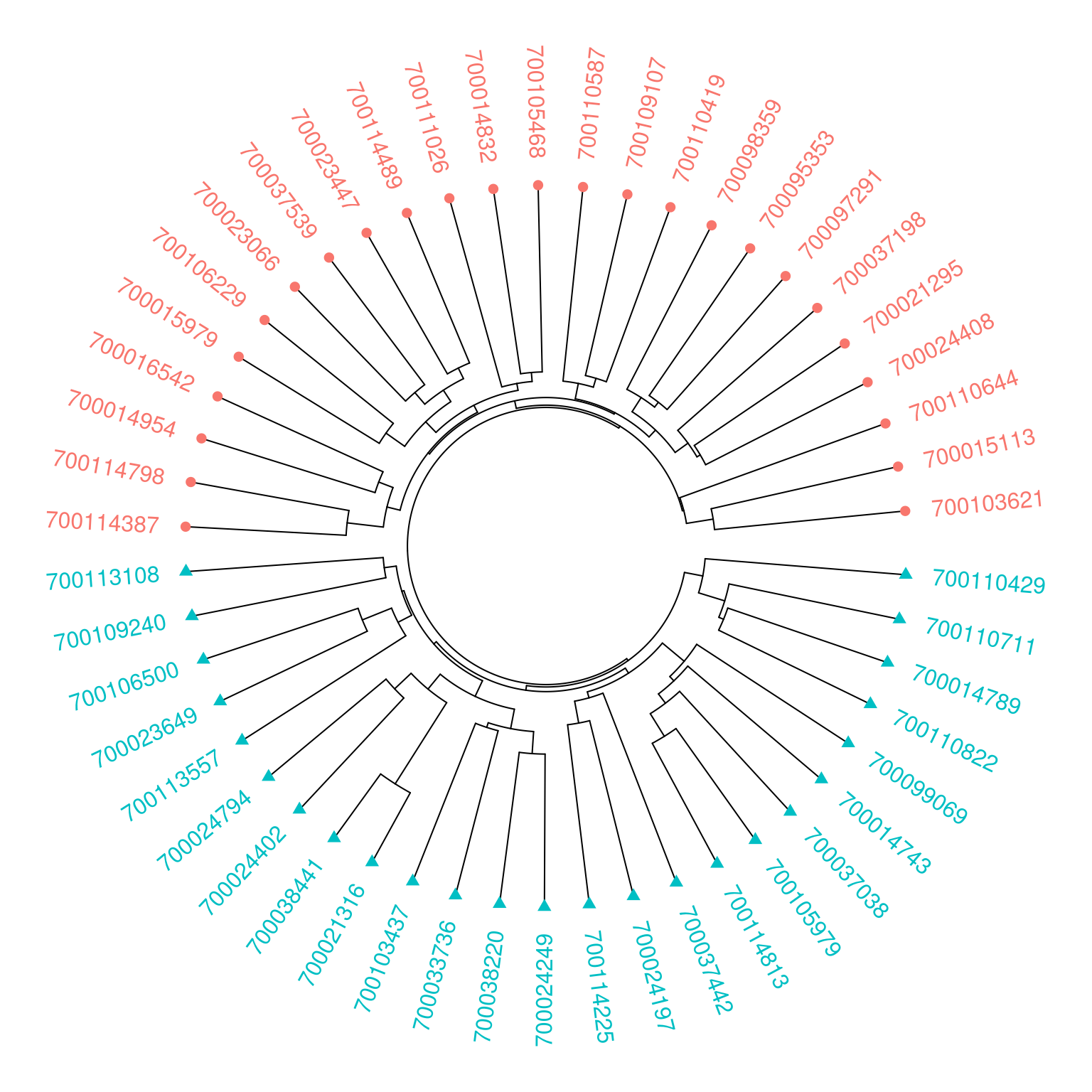
Principle Coordinates Analysis
The phyloseq
package additionally provides methods for commonly-used ordination
analyses such as principle coordinates analysis. The
ordinate method simply requires a phyloseq
object and specification of the type of ordination analysis to be
preformed. The type of distance method used can also optionally be
specified.
V13_ordination <-
ordinate(V13_phyloseq, method = "PCoA", distance = "bray")The ordination analysis can then be plotted using the
plot_ordination method provided by the phyloseq
package. Again, additional ggplot2
syntax is used to control the presentational aspects of the plot.
V13_phyloseq %>%
plot_ordination(V13_ordination, color = "Study", shape = "Study") +
theme_bw() +
theme(legend.position = "bottom")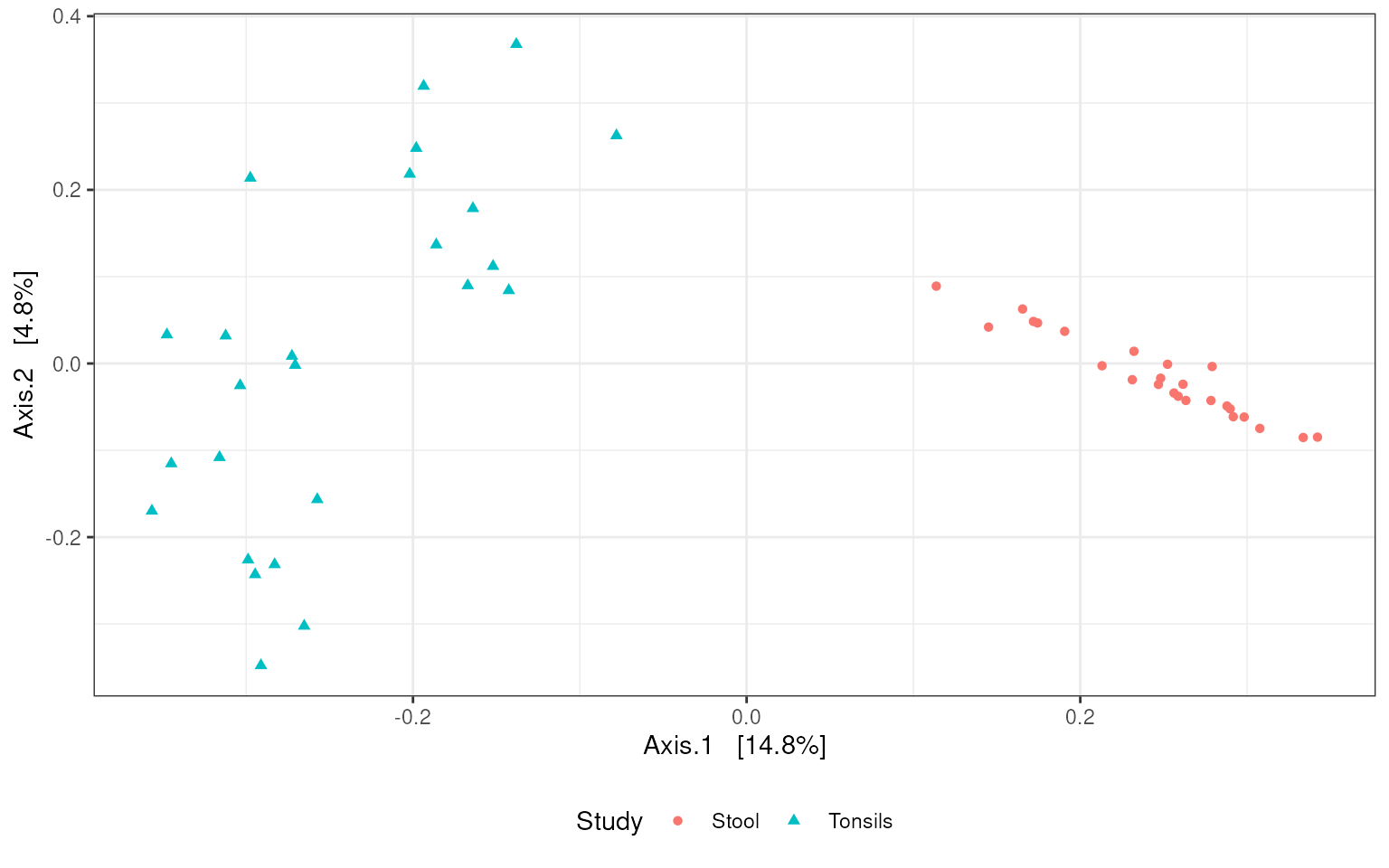
Finally, the ordination eigenvalues can be plotted using the
plot_scree method provided by the phyloseq
package.
V13_ordination %>%
plot_scree() +
theme_bw()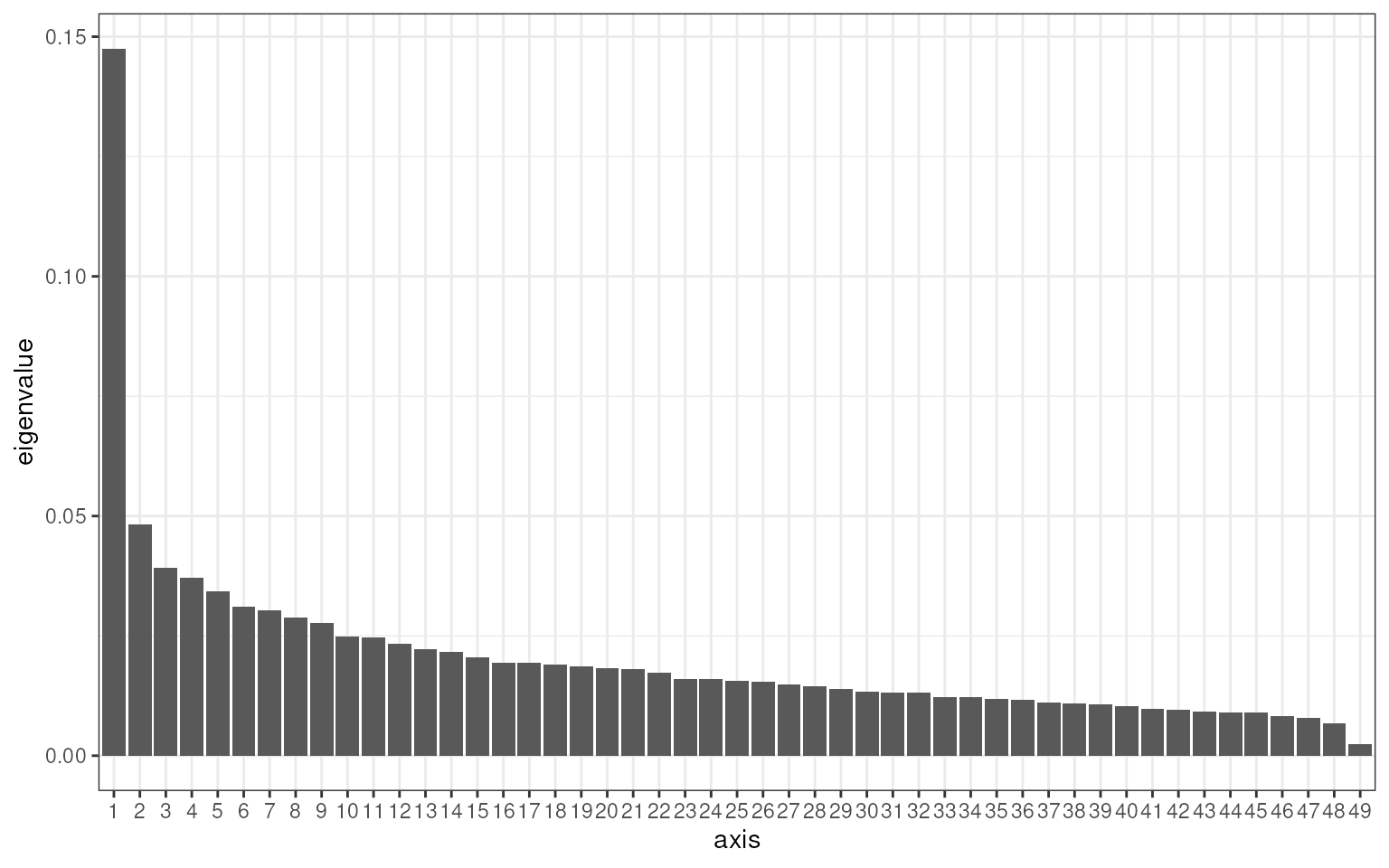
Phylum-level Comparison to Metagenomic Shotgun Sequencing
In addition to 16S rRNA sequencing, the HMP Study conducted whole metagenome shotgun (MGX) sequencing. These profiles, along with thousands of profiles from other studies, are available in the curatedMetagenomicData package. Here, a phylum-level relative abundance comparison of the 16S and MGX samples is made to illustrate comparing these data sets.
First a V35_stool_phyloseq object is constructed and
contains the 16S variable region 3–5 data for stool samples. Then the
MGX stool samples are obtained and coerced to a phyloseq
object as follows.
V35_stool_phyloseq <-
V35_stool %>%
as_phyloseq()
EH <-
ExperimentHub()
HMP_2012.metaphlan_bugs_list.stool <-
EH[["EH426"]]
# a modified version of ExpressionSet2phyloseq taken from curatedMetagenomicData
# ExpressionSet2phyloseq was removed in curatedMetagenomicData version 3.0.0
ExpressionSet2phyloseq <- function(eset, simplify = TRUE, relab = TRUE) {
otu.table <-
Biobase::exprs(eset)
sample.data <-
Biobase::pData(eset) %>%
phyloseq::sample_data(., errorIfNULL = FALSE)
taxonomic.ranks <-
c("Kingdom", "Phylum", "Class", "Order", "Family", "Genus", "Species", "Strain")
tax.table <-
rownames(otu.table) %>%
gsub("[a-z]__", "", .) %>%
dplyr::tibble() %>%
tidyr::separate(., ".", taxonomic.ranks, sep = "\\|", fill="right") %>%
as.matrix()
if(simplify) {
rownames(otu.table) <- rownames(otu.table) %>%
gsub(".+\\|", "", .)
}
rownames(tax.table) <-
rownames(otu.table)
if(!relab) {
otu.table <-
round(sweep(otu.table, 2, eset$number_reads/100, "*"))
}
otu.table <-
phyloseq::otu_table(otu.table, taxa_are_rows = TRUE)
tax.table <-
phyloseq::tax_table(tax.table)
phyloseq::phyloseq(otu.table, sample.data, tax.table)
}
MGX_stool_phyloseq <-
ExpressionSet2phyloseq(HMP_2012.metaphlan_bugs_list.stool)The curatedMetagenomicData package provides taxonomic relative abundance for MGX data (with an option to estimate counts by multiplying by read depth) from MetaPhlAn2. MetaPhlAn2 directly estimates relative abundance at every taxonomic level based on clade-specific markers, and these estimates are better than summing lower-level taxonomic abundances.
Note, this comparison becomes complicated because phyloseq
does not currently support row and column matching and reordering.
Instead, we use the phyloseq::psmelt() method to generate
data.frame objects for further manipulation.
The 16S data sets do not contain summarized counts for higher
taxonomic levels, so we use the phyloseq::tax_glom() method
to agglomerate at phylum level.
There is a column SRS_SAMPLE_ID present in the 16S
variable region 3–5 data that is explicitly for mapping to the MGX
samples and the matching identifiers are in the
NCBI_accession column of the MGX sample data. The
intersection of the two vectors represents the matching samples that are
present in both data sets. This intersection, shown below as
SRS_SAMPLE_IDS, can then be used to filter both the 16S and
MGX samples to include only the samples in common.
Along with the filter step shown below, standardization
of sample identifiers to the SRS numbers and conversion of
taxonomic counts to relative abundance is done. The conversion to
relative abundance is necessary for comparability across the 16S and MGX
samples.
In either case, data is first grouped by samples and the percent composition of each phylum relative to the others in the sample is calculated by taking the count abundance of the phylum divided by the sum of all abundance counts in the sample. Samples are then sorted by phylum and descending relative abundance before being grouped by phylum – the grouping by phylum allows for the assignment a phylum rank that is then used to sort the phyla by descending relative abundance. Finally, only the sample, phylum, and relative abundance columns are kept once the order has been established and all groupings can be removed.
SRS_SAMPLE_IDS <-
intersect(V35_stool_melted$SRS_SAMPLE_ID, MGX_stool_melted$NCBI_accession)
V35_stool_melted %<>%
filter(SRS_SAMPLE_ID %in% SRS_SAMPLE_IDS) %>%
rename(Phylum = PHYLUM) %>%
mutate(Sample = SRS_SAMPLE_ID) %>%
group_by(Sample) %>%
mutate(`Relative Abundance` = Abundance / sum(Abundance)) %>%
arrange(Phylum, -`Relative Abundance`) %>%
group_by(Phylum) %>%
mutate(phylum_rank = sum(`Relative Abundance`)) %>%
arrange(-phylum_rank) %>%
select(Sample, Phylum, `Relative Abundance`) %>%
ungroup()
MGX_stool_melted %<>%
filter(NCBI_accession %in% SRS_SAMPLE_IDS) %>%
group_by(Sample) %>%
mutate(`Relative Abundance` = Abundance / sum(Abundance)) %>%
arrange(Phylum, -`Relative Abundance`) %>%
group_by(Phylum) %>%
mutate(phylum_rank = sum(`Relative Abundance`)) %>%
arrange(-phylum_rank) %>%
select(Sample, Phylum, `Relative Abundance`) %>%
ungroup()Provided that the phyla are ordered by relative abundance, the top phyla from each data set can be obtained and intersected to give the top eight phyla present in both data sets. The top eight phyla can then be used to filter 16S and MGX samples to include only the desired phyla.
V35_top_phyla <-
V35_stool_melted %$%
unique(Phylum) %>%
as.character()
MGX_top_phyla <-
MGX_stool_melted %$%
unique(Phylum) %>%
as.character()
top_eight_phyla <-
intersect(V35_top_phyla, MGX_top_phyla) %>%
extract(1:8)
V35_stool_melted %<>%
filter(Phylum %in% top_eight_phyla)
MGX_stool_melted %<>%
filter(Phylum %in% top_eight_phyla)To achieve ordering of samples by the relative abundance of the top
phylum when plotting, a vector of the unique sample identifiers is
constructed and will be used as the levels of a
factor.
A vector of unique phyla is also constructed and will be used as the
levels of a factor when plotting. If this were
not done, phyla would simply be sorted alphabetically.
Color blindness affects a significant portion of the population, but, using an intelligent color pallet, figures can be designed to be friendly to those with deuteranopia, protanopia, and tritanopia. The following colors are derived from Wong, B. Points of view: Color blindness. Nat. Methods 8, 441 (2011).
bang_wong_colors <-
c("#CC79A7", "#D55E00", "#0072B2", "#F0E442", "#009E73", "#56B4E9",
"#E69F00", "#000000")Using the sample_order and phylum_order
vectors constructed above, stacked phylum-level relative abundance bar
plots sorted by decreasing relative abundance of the top phylum and
stratified by the top eight phyla can be made for 16S and MGX samples.
The two plots are made separately and serialized so that they can be
arranged in a single figure for comparison.
V35_plot <-
V35_stool_melted %>%
mutate(Sample = factor(Sample, sample_order)) %>%
mutate(Phylum = factor(Phylum, phylum_order)) %>%
ggplot(aes(Sample, `Relative Abundance`, fill = Phylum)) +
geom_bar(stat = "identity", position = "fill", width = 1) +
scale_fill_manual(values = bang_wong_colors) +
theme_minimal() +
theme(axis.text.x = element_blank(), axis.title.x = element_blank(),
panel.grid = element_blank(), legend.position = "none",
legend.title = element_blank()) +
ggtitle("Phylum-Level Relative Abundance", "16S Stool Samples")
MGX_plot <-
MGX_stool_melted %>%
mutate(Sample = factor(Sample, sample_order)) %>%
mutate(Phylum = factor(Phylum, phylum_order)) %>%
ggplot(aes(Sample, `Relative Abundance`, fill = Phylum)) +
geom_bar(stat = "identity", position = "fill", width = 1) +
scale_fill_manual(values = bang_wong_colors) +
theme_minimal() +
theme(axis.text.x = element_blank(), axis.title.x = element_blank(),
panel.grid = element_blank(), legend.position = "none",
legend.title = element_blank()) +
ggtitle("", "MGX Stool Samples")In the plots above, legends are removed to reduce redundancy, but a
legend is still necessary and can be serialized as its own plot using
the get_legend method from the cowplot
package.
plot_legend <- {
MGX_plot +
theme(legend.position = "bottom")
} %>%
get_legend()Finally, the grid.arrange method from the gridExtra
package is used to arrange, scale, and plot the three plots in a single
figure.
grid.arrange(V35_plot, MGX_plot, plot_legend, ncol = 1, heights = c(3, 3, 1))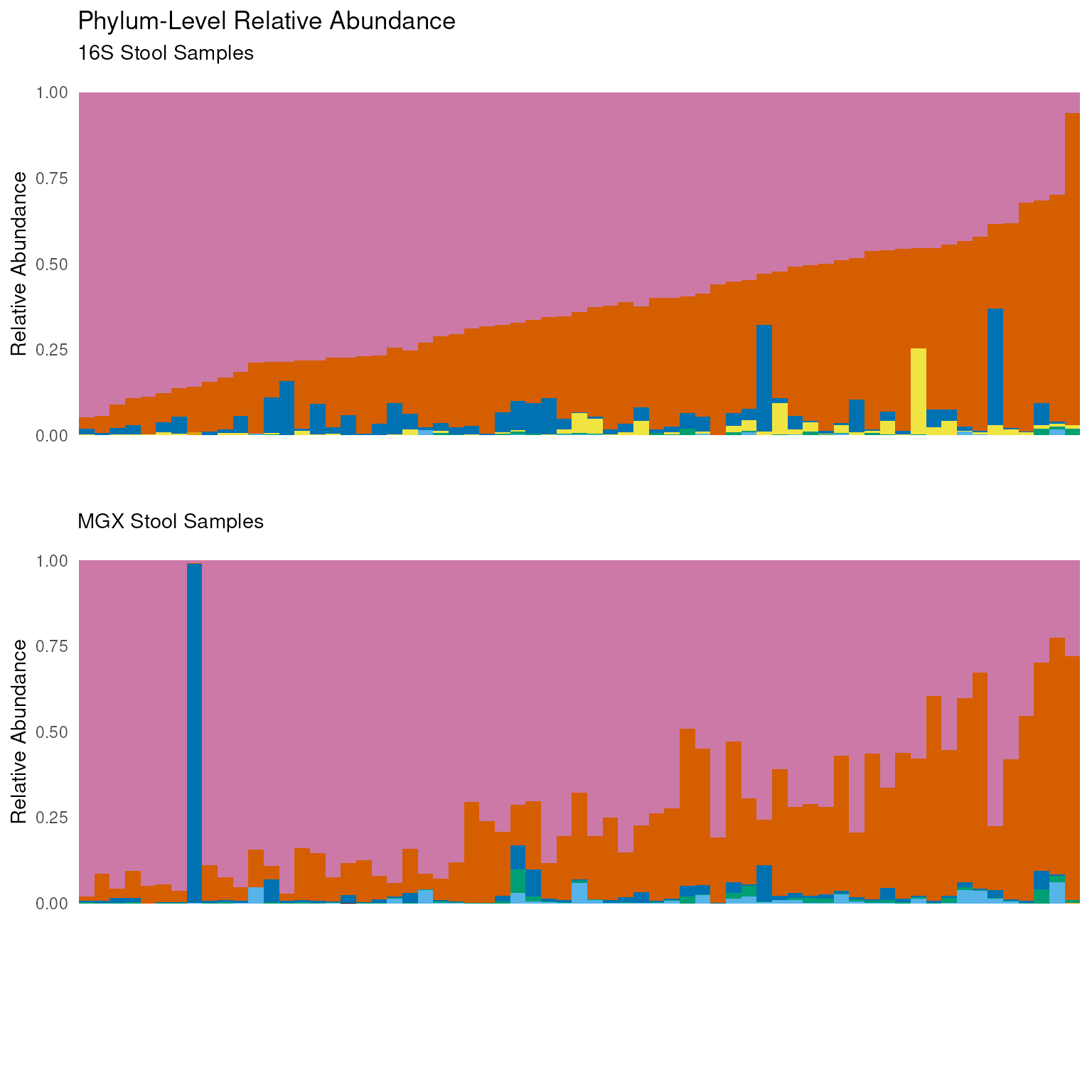
Notably, the figure illustrates the Bacteroidetes/Firmicutes gradient with reasonable agreement between the 16S and MGX samples.
When these plots were submitted for publication, the following code was used to produce ESP and PDF files.
V35_plot +
theme(text = element_text(size = 19)) +
labs(title = NULL, subtitle = NULL, tag = "A")
ggsave("~/AJE-00611-2018 Schiffer Figure 2A.eps", device = "eps")
ggsave("~/AJE-00611-2018 Schiffer Figure 2A.pdf", device = "pdf")
MGX_plot +
theme(text = element_text(size = 19)) +
labs(title = NULL, subtitle = NULL, tag = "B")
ggsave("~/AJE-00611-2018 Schiffer Figure 2B.eps", device = "eps")
ggsave("~/AJE-00611-2018 Schiffer Figure 2B.pdf", device = "pdf")
plot_legend <- {
MGX_plot +
theme(legend.position = "bottom", text = element_text(size = 19)) +
guides(fill = guide_legend(byrow = TRUE))
} %>%
get_legend()
ggsave("~/AJE-00611-2018 Schiffer Figure 2 Legend 1.eps", plot = plot_legend,
device = "eps")
ggsave("~/AJE-00611-2018 Schiffer Figure 2 Legend 1.pdf", plot = plot_legend,
device = "pdf")
plot_legend <- {
MGX_plot +
theme(legend.position = "right", text = element_text(size = 19))
} %>%
get_legend()
ggsave("~/AJE-00611-2018 Schiffer Figure 2 Legend 2.eps", plot = plot_legend,
device = "eps")
ggsave("~/AJE-00611-2018 Schiffer Figure 2 Legend 2.pdf", plot = plot_legend,
device = "pdf")
plot_legend <- {
MGX_plot +
theme(legend.position = "right", text = element_text(size = 19)) +
guides(fill = guide_legend(ncol = 2, byrow = TRUE))
} %>%
get_legend()
ggsave("~/AJE-00611-2018 Schiffer Figure 2 Legend 3.eps", plot = plot_legend,
device = "eps")
ggsave("~/AJE-00611-2018 Schiffer Figure 2 Legend 3.pdf", plot = plot_legend,
device = "pdf")
V35_plot <-
V35_plot +
theme(text = element_text(size = 19)) +
labs(title = NULL, subtitle = NULL, tag = "A")
MGX_plot <-
MGX_plot +
theme(text = element_text(size = 19)) +
labs(title = NULL, subtitle = NULL, tag = "B")
plot_legend <- {
MGX_plot +
theme(legend.position = "bottom", text = element_text(size = 19)) +
guides(fill = guide_legend(byrow = TRUE))
} %>%
get_legend()
grid_object <-
grid.arrange(V35_plot, MGX_plot, plot_legend, ncol = 1,
heights = c(3, 3, 1))
ggsave("~/AJE-00611-2018 Schiffer Figure 3.eps", plot = grid_object,
device = "eps", width = 8, height = 8)
ggsave("~/AJE-00611-2018 Schiffer Figure 3.pdf", plot = grid_object,
device = "pdf", width = 8, height = 8)Exporting Data to CSV, SAS, SPSS, and STATA Formats
To our knowledge, R and Bioconductor provide the most and best methods for the analysis of microbiome data. However, we realize they are not the only analysis environments and wish to provide methods to export the data from HMP16SData to CSV, SAS, SPSS, and STATA formats. As we do not use these other languages regularly, we are unaware of how to represent phylogenitic trees in them and will not attempt to export the trees here.
Prepare Data for Export
Bioconductor’s SummarizedExperiment class is essentially
a normalized representation of tightly coupled data and metadata that
must be “unglued” before it can be saved into alternative formats.
The process begins by creating a data.frame object of
the participant data and moving the row names to their own column.
V13_participant_data <-
V13() %>%
colData() %>%
as.data.frame() %>%
rownames_to_column(var = "PSN")Next, the taxonomic abundance matrix is extracted and transposed
because Bioconductor objects represent samples as rows and measurements
as columns whereas other languages do the opposite. The matrix is then
coerced to a data.frame object and the row names are moved
to their own column.
V13_OTU_counts <-
V13() %>%
assay() %>%
t() %>%
as.data.frame() %>%
rownames_to_column(var = "PSN")With the participant data and taxonomic abundances represented as
simple tables containing a primary key (i.e. PSN or Primary Sample
Number) the two tables can be joined using the
merge.data.frame method.
V13_data <-
merge.data.frame(V13_participant_data, V13_OTU_counts, by = "PSN")The column names of the V13_data object are denoted as
OTUs or operational taxonomic units based on their sequence similarity
to the 16S rRNA gene. The OTU nomenclature is not particularly useful
without the traditional taxonomic clade identifiers which are stored in
a separate table. A dictionary of these identifiers is created by
extracting and transposing the rowData of the
SummarizedExperiment object which is then coerced to a
data.frame object.
V13_dictionary <-
V13() %>%
rowData() %>%
t.data.frame() %>%
as.data.frame()The column names of the V13_data object contain periods
which some languages and formats are unable to process. The periods in
the column names are replaced with underscores using the
gsub method.
The process is repeated for the column names of the
V13_dictionary object.
colnames(V13_dictionary) <-
colnames(V13_dictionary) %>%
gsub(pattern = "\\.", replacement = "_", x = .)The two tables V13_data and V13_dictionary
are then ready for export to CSV, SAS, SPSS, and STATA formats.
Export to CSV Format
To export to CSV format, two calls to the write_csv
method from the readr package
are used to write CSV files to disk.
Export to SAS Format
To export to SAS format, two calls to the write_sas
method from the haven package
are used to write SAS files to disk.
Export to SPSS Format
To export to SPSS format, two calls to the write_sav
method from the haven package
are used to write SPSS files to disk.
Export to STATA Format
To export to STATA format, two calls to the write_dta
method from the haven package
are used to write STATA files to disk.
write_dta(V13_data, "~/V13_data.dta", version = 13)
write_dta(V13_dictionary, "~/V13_dictionary.dta", version = 13)Here, version 13 STATA files are written because version 14 and above files require a platform-specific text encoding that would make the data less transportable. The encoding of the version 13 files is ASCII.
Session Information
## R Under development (unstable) (2026-03-22 r89674)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: Etc/UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] haven_2.5.5 readr_2.2.0
## [3] cowplot_1.2.0 gridExtra_2.3
## [5] ExperimentHub_3.1.0 AnnotationHub_4.1.0
## [7] BiocFileCache_3.1.0 dbplyr_2.5.2
## [9] circlize_0.4.17 dendextend_1.19.1
## [11] dplyr_1.2.0 tibble_3.3.1
## [13] ggplot2_4.0.2 magrittr_2.0.4
## [15] phyloseq_1.55.2 HMP16SData_1.30.0
## [17] SummarizedExperiment_1.41.1 Biobase_2.71.0
## [19] GenomicRanges_1.63.1 Seqinfo_1.1.0
## [21] IRanges_2.45.0 S4Vectors_0.49.0
## [23] BiocGenerics_0.57.0 generics_0.1.4
## [25] MatrixGenerics_1.23.0 matrixStats_1.5.0
## [27] BiocStyle_2.39.0
##
## loaded via a namespace (and not attached):
## [1] DBI_1.3.0 httr2_1.2.2 permute_0.9-10
## [4] rlang_1.1.7 ade4_1.7-24 otel_0.2.0
## [7] compiler_4.6.0 RSQLite_2.4.6 mgcv_1.9-4
## [10] png_0.1-9 systemfonts_1.3.2 vctrs_0.7.2
## [13] reshape2_1.4.5 stringr_1.6.0 shape_1.4.6.1
## [16] pkgconfig_2.0.3 crayon_1.5.3 fastmap_1.2.0
## [19] XVector_0.51.0 labeling_0.4.3 rmarkdown_2.30
## [22] tzdb_0.5.0 ragg_1.5.2 purrr_1.2.1
## [25] bit_4.6.0 xfun_0.57 cachem_1.1.0
## [28] jsonlite_2.0.0 biomformat_1.39.10 blob_1.3.0
## [31] DelayedArray_0.37.0 cluster_2.1.8.2 parallel_4.6.0
## [34] R6_2.6.1 bslib_0.10.0 stringi_1.8.7
## [37] RColorBrewer_1.1-3 jquerylib_0.1.4 Rcpp_1.1.1
## [40] bookdown_0.46 assertthat_0.2.1 iterators_1.0.14
## [43] knitr_1.51 splines_4.6.0 igraph_2.2.2
## [46] Matrix_1.7-5 tidyselect_1.2.1 viridis_0.6.5
## [49] rstudioapi_0.18.0 abind_1.4-8 yaml_2.3.12
## [52] vegan_2.7-3 codetools_0.2-20 curl_7.0.0
## [55] lattice_0.22-9 plyr_1.8.9 S7_0.2.1
## [58] withr_3.0.2 KEGGREST_1.51.1 evaluate_1.0.5
## [61] survival_3.8-6 desc_1.4.3 xml2_1.5.2
## [64] Biostrings_2.79.5 pillar_1.11.1 BiocManager_1.30.27
## [67] filelock_1.0.3 foreach_1.5.2 BiocVersion_3.23.1
## [70] hms_1.1.4 scales_1.4.0 glue_1.8.0
## [73] tools_4.6.0 data.table_1.18.2.1 forcats_1.0.1
## [76] fs_2.0.1 grid_4.6.0 tidyr_1.3.2
## [79] ape_5.8-1 colorspace_2.1-2 AnnotationDbi_1.73.0
## [82] nlme_3.1-168 cli_3.6.5 rappdirs_0.3.4
## [85] kableExtra_1.4.0 textshaping_1.0.5 S4Arrays_1.11.1
## [88] viridisLite_0.4.3 svglite_2.2.2 gtable_0.3.6
## [91] sass_0.4.10 digest_0.6.39 SparseArray_1.11.11
## [94] htmlwidgets_1.6.4 farver_2.1.2 multtest_2.67.0
## [97] memoise_2.0.1 htmltools_0.5.9 pkgdown_2.2.0
## [100] lifecycle_1.0.5 httr_1.4.8 GlobalOptions_0.1.3
## [103] bit64_4.6.0-1 MASS_7.3-65