PublicDataResources.RmdInstallation
If you are using the Docker container provided with this workshop, everything (including the workshop) is already installed. Otherwise, you will need to install the following packages.
Sean Davis
GEOquery
(Davis and Meltzer 2007)
The NCBI Gene Expression Omnibus (GEO) serves as a public repository for a wide range of high-throughput experimental data. These data include single and dual channel microarray-based experiments measuring mRNA, genomic DNA, and protein abundance, as well as non-array techniques such as serial analysis of gene expression (SAGE), mass spectrometry proteomic data, and high-throughput sequencing data. The GEOquery package (Davis and Meltzer 2007) forms a bridge between this public repository and the analysis capabilities in Bioconductor.
Overview of GEO
At the most basic level of organization of GEO, there are four basic entity types. The first three (Sample, Platform, and Series) are supplied by users; the fourth, the dataset, is compiled and curated by GEO staff from the user-submitted data. See the GEO home page for more information.
Platforms
A Platform record describes the list of elements on the array (e.g., cDNAs, oligonucleotide probesets, ORFs, antibodies) or the list of elements that may be detected and quantified in that experiment (e.g., SAGE tags, peptides). Each Platform record is assigned a unique and stable GEO accession number (GPLxxx). A Platform may reference many Samples that have been submitted by multiple submitters.
Samples
A Sample record describes the conditions under which an individual Sample was handled, the manipulations it underwent, and the abundance measurement of each element derived from it. Each Sample record is assigned a unique and stable GEO accession number (GSMxxx). A Sample entity must reference only one Platform and may be included in multiple Series.
Series
A Series record defines a set of related Samples considered to be part of a group, how the Samples are related, and if and how they are ordered. A Series provides a focal point and description of the experiment as a whole. Series records may also contain tables describing extracted data, summary conclusions, or analyses. Each Series record is assigned a unique and stable GEO accession number (GSExxx). Series records are available in a couple of formats which are handled by GEOquery independently. The smaller and new GSEMatrix files are quite fast to parse; a simple flag is used by GEOquery to choose to use GSEMatrix files (see below).
Getting Started using GEOquery
Getting data from GEO is really quite easy. There is only one command that is needed, getGEO. This one function interprets its input to determine how to get the data from GEO and then parse the data into useful R data structures.
With the library loaded, we are free to access any GEO accession.
Use case: MDS plot of cancer data
The data we are going to access are from this paper.
Background: The tumor microenvironment is an important factor in cancer immunotherapy response. To further understand how a tumor affects the local immune system, we analyzed immune gene expression differences between matching normal and tumor tissue.Methods: We analyzed public and new gene expression data from solid cancers and isolated immune cell populations. We also determined the correlation between CD8, FoxP3 IHC, and our gene signatures.Results: We observed that regulatory T cells (Tregs) were one of the main drivers of immune gene expression differences between normal and tumor tissue. A tumor-specific CD8 signature was slightly lower in tumor tissue compared with normal of most (12 of 16) cancers, whereas a Treg signature was higher in tumor tissue of all cancers except liver. Clustering by Treg signature found two groups in colorectal cancer datasets. The high Treg cluster had more samples that were consensus molecular subtype 1/4, right-sided, and microsatellite-instable, compared with the low Treg cluster. Finally, we found that the correlation between signature and IHC was low in our small dataset, but samples in the high Treg cluster had significantly more CD8+ and FoxP3+ cells compared with the low Treg cluster.Conclusions: Treg gene expression is highly indicative of the overall tumor immune environment.Impact: In comparison with the consensus molecular subtype and microsatellite status, the Treg signature identifies more colorectal tumors with high immune activation that may benefit from cancer immunotherapy.
In this little exercise, we will:
- Access public omics data using the GEOquery package
- Convert the public omics data to a
SummarizedExperimentobject. - Perform a simple unsupervised analysis to visualize these public data.
Use the GEOquery package to fetch data about GSE103512.
gse = getGEO("GSE103512")[[1]]## Warning: 102 parsing failures.
## row col expected actual file
## 54614 SPOT_ID 1/0/T/F/TRUE/FALSE --Control literal data
## 54615 SPOT_ID 1/0/T/F/TRUE/FALSE --Control literal data
## 54616 SPOT_ID 1/0/T/F/TRUE/FALSE --Control literal data
## 54617 SPOT_ID 1/0/T/F/TRUE/FALSE --Control literal data
## 54618 SPOT_ID 1/0/T/F/TRUE/FALSE --Control literal data
## ..... ....... .................. ......... ............
## See problems(...) for more details.Note that getGEO, when used to retrieve GSE records, returns a list. The members of the list each represent one GEO Platform, since each GSE record can contain multiple related datasets (eg., gene expression and DNA methylation). In this case, the list is of length one, but it is still necessary to grab the first elment.
The first step–a detail–is to convert from the older Bioconductor data structure (GEOquery was written in 2007), the ExpressionSet, to the newer SummarizedExperiment. One line suffices.
library(SummarizedExperiment)
se = as(gse, "SummarizedExperiment")Examine two variables of interest, cancer type and tumor/normal status.
## normal.ch1
## cancer.type.ch1 no yes
## BC 65 10
## CRC 57 12
## NSCLC 60 9
## PCA 60 7Filter gene expression by variance to find most informative genes.
Perform multidimensional scaling and prepare for plotting. We will be using ggplot2, so we need to make a data.frame before plotting.
mdsvals = cmdscale(dist(t(dat)))
mdsvals = as.data.frame(mdsvals)
mdsvals$Type=factor(colData(se)[,'cancer.type.ch1'])
mdsvals$Normal = factor(colData(se)[,'normal.ch1'])
head(mdsvals)## V1 V2 Type Normal
## GSM2772660 8.531331 18.57115 BC no
## GSM2772661 8.991591 13.63764 BC no
## GSM2772662 10.788973 13.48403 BC no
## GSM2772663 3.127105 19.13529 BC no
## GSM2772664 13.056599 13.88711 BC no
## GSM2772665 7.903717 13.24731 BC noAnd do the plot.
library(ggplot2)
ggplot(mdsvals, aes(x=V1,y=V2,shape=Normal,color=Type)) +
geom_point( alpha=0.6) + theme(text=element_text(size = 18))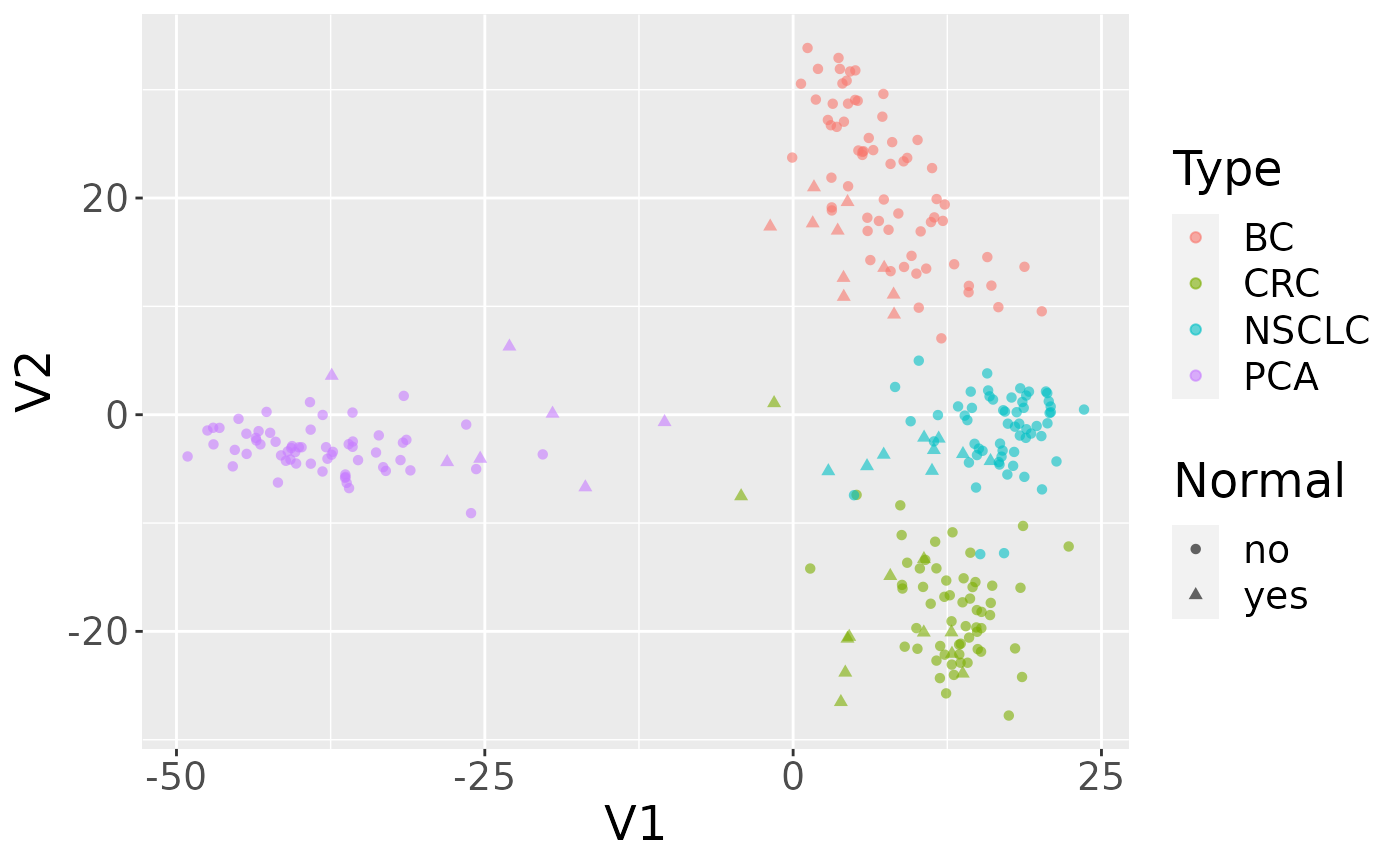
Accessing Raw Data from GEO
NCBI GEO accepts (but has not always required) raw data such as .CEL files, .CDF files, images, etc. It is also not uncommon for some RNA-seq or other sequencing datasets to supply only raw data (with accompanying sample information, of course), necessitating Sometimes, it is useful to get quick access to such data. A single function, getGEOSuppFiles, can take as an argument a GEO accession and will download all the raw data associate with that accession. By default, the function will create a directory in the current working directory to store the raw data for the chosen GEO accession.
GenomicDataCommons
From the Genomic Data Commons (GDC) website:
The National Cancer Institute’s (NCI’s) Genomic Data Commons (GDC) is a data sharing platform that promotes precision medicine in oncology. It is not just a database or a tool; it is an expandable knowledge network supporting the import and standardization of genomic and clinical data from cancer research programs. The GDC contains NCI-generated data from some of the largest and most comprehensive cancer genomic datasets, including The Cancer Genome Atlas (TCGA) and Therapeutically Applicable Research to Generate Effective Therapies (TARGET). For the first time, these datasets have been harmonized using a common set of bioinformatics pipelines, so that the data can be directly compared. As a growing knowledge system for cancer, the GDC also enables researchers to submit data, and harmonizes these data for import into the GDC. As more researchers add clinical and genomic data to the GDC, it will become an even more powerful tool for making discoveries about the molecular basis of cancer that may lead to better care for patients.
The data model for the GDC is complex, but it worth a quick overview and a graphical representation is included here.
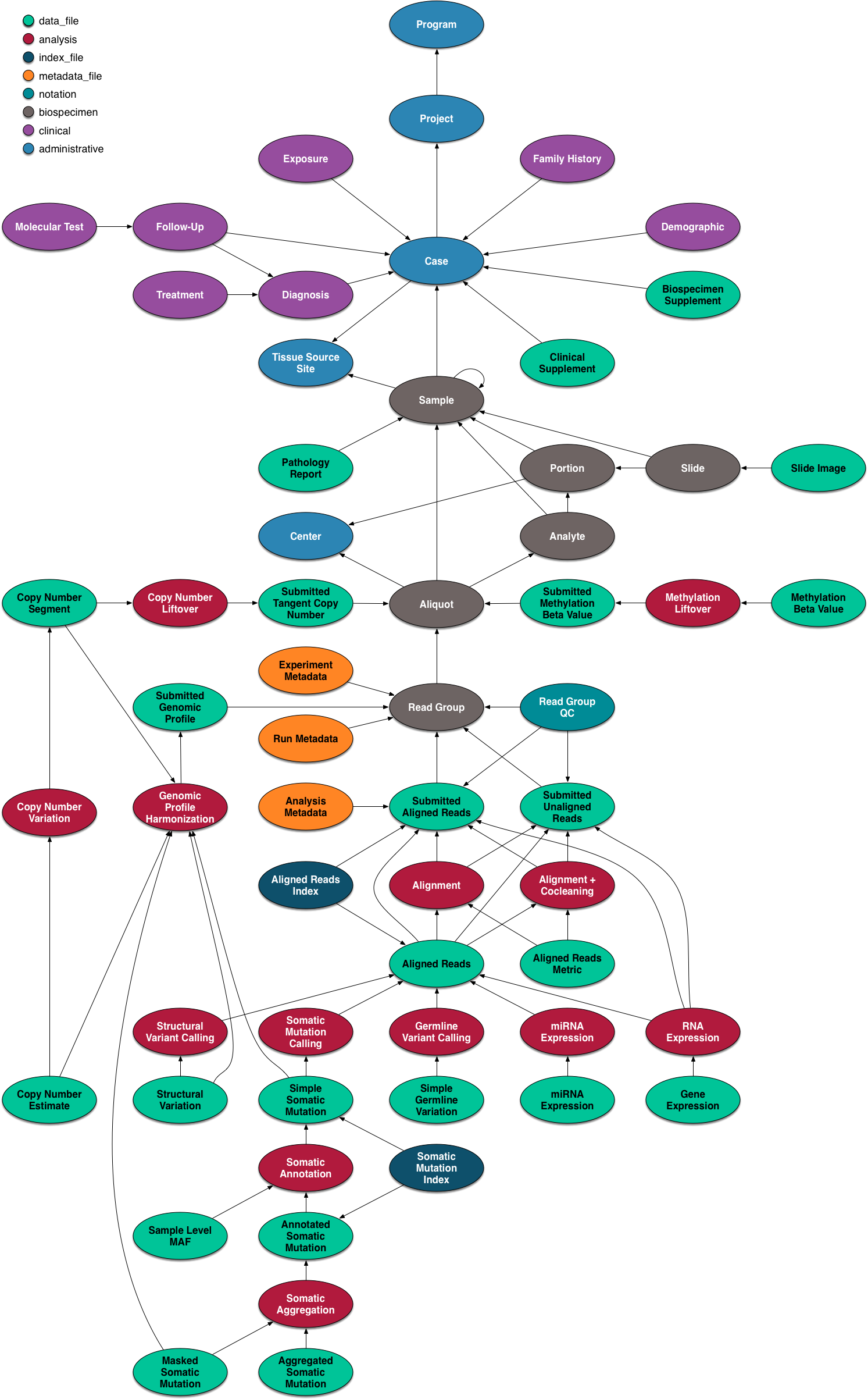
The data model is encoded as a so-called property graph. Nodes represent entities such as Projects, Cases, Diagnoses, Files (various kinds), and Annotations. The relationships between these entities are maintained as edges. Both nodes and edges may have Properties that supply instance details.
The GDC API exposes these nodes and edges in a somewhat simplified set of RESTful endpoints.
Quickstart
This quickstart section is just meant to show basic functionality. More details of functionality are included further on in this vignette and in function-specific help.
To report bugs or problems, either submit a new issue or submit a bug.report(package='GenomicDataCommons') from within R (which will redirect you to the new issue on GitHub).
Check connectivity and status
The GenomicDataCommons package relies on having network connectivity. In addition, the NCI GDC API must also be operational and not under maintenance. Checking status can be used to check this connectivity and functionality.
GenomicDataCommons::status()## $commit
## [1] "dfa394478bd39c11b89d3819a398898d99575a24"
##
## $data_release
## [1] "Data Release 29.0 - March 31, 2021"
##
## $status
## [1] "OK"
##
## $tag
## [1] "3.0.0"
##
## $version
## [1] 1Find data
The following code builds a manifest that can be used to guide the download of raw data. Here, filtering finds gene expression files quantified as raw counts using HTSeq from ovarian cancer patients.
Download data
After the 379 gene expression files specified in the query above. Using multiple processes to do the download very significantly speeds up the transfer in many cases. On a standard 1Gb connection, the following completes in about 30 seconds. The first time the data are downloaded, R will ask to create a cache directory (see ?gdc_cache for details of setting and interacting with the cache). Resulting downloaded files will be stored in the cache directory. Future access to the same files will be directly from the cache, alleviating multiple downloads.
fnames = lapply(ge_manifest$id[1:20],gdcdata)If the download had included controlled-access data, the download above would have needed to include a token. Details are available in the authentication section below.
Metadata queries
The GenomicDataCommons can access the significant clinical, demographic, biospecimen, and annotation information contained in the NCI GDC.
expands = c("diagnoses","annotations",
"demographic","exposures")
projResults = projects() %>%
results(size=10)
str(projResults,list.len=5)## List of 9
## $ id : chr [1:10] "GENIE-MSK" "TCGA-UCEC" "TCGA-LGG" "TCGA-SARC" ...
## $ primary_site :List of 10
## ..$ GENIE-MSK : chr [1:49] "Testis" "Gallbladder" "Unknown" "Other and unspecified parts of biliary tract" ...
## ..$ TCGA-UCEC : chr [1:2] "Corpus uteri" "Uterus, NOS"
## ..$ TCGA-LGG : chr "Brain"
## ..$ TCGA-SARC : chr [1:13] "Kidney" "Other and unspecified parts of tongue" "Bones, joints and articular cartilage of limbs" "Colon" ...
## ..$ TCGA-PAAD : chr "Pancreas"
## .. [list output truncated]
## $ dbgap_accession_number: logi [1:10] NA NA NA NA NA NA ...
## $ project_id : chr [1:10] "GENIE-MSK" "TCGA-UCEC" "TCGA-LGG" "TCGA-SARC" ...
## $ disease_type :List of 10
## ..$ GENIE-MSK : chr [1:49] "Germ Cell Neoplasms" "Granular Cell Tumors and Alveolar Soft Part Sarcomas" "Immunoproliferative Diseases" "Plasma Cell Tumors" ...
## ..$ TCGA-UCEC : chr [1:4] "Epithelial Neoplasms, NOS" "Cystic, Mucinous and Serous Neoplasms" "Adenomas and Adenocarcinomas" "Not Reported"
## ..$ TCGA-LGG : chr "Gliomas"
## ..$ TCGA-SARC : chr [1:6] "Nerve Sheath Tumors" "Myomatous Neoplasms" "Fibromatous Neoplasms" "Lipomatous Neoplasms" ...
## ..$ TCGA-PAAD : chr [1:4] "Cystic, Mucinous and Serous Neoplasms" "Ductal and Lobular Neoplasms" "Adenomas and Adenocarcinomas" "Epithelial Neoplasms, NOS"
## .. [list output truncated]
## [list output truncated]
## - attr(*, "row.names")= int [1:10] 1 2 3 4 5 6 7 8 9 10
## - attr(*, "class")= chr [1:3] "GDCprojectsResults" "GDCResults" "list"
names(projResults)## [1] "id" "primary_site" "dbgap_accession_number"
## [4] "project_id" "disease_type" "name"
## [7] "releasable" "state" "released"
# or listviewer::jsonedit(clinResults)Basic design
This package design is meant to have some similarities to the “hadleyverse” approach of dplyr. Roughly, the functionality for finding and accessing files and metadata can be divided into:
- Simple query constructors based on GDC API endpoints.
- A set of verbs that when applied, adjust filtering, field selection, and faceting (fields for aggregation) and result in a new query object (an endomorphism)
- A set of verbs that take a query and return results from the GDC
In addition, there are exhiliary functions for asking the GDC API for information about available and default fields, slicing BAM files, and downloading actual data files. Here is an overview of functionality1.
- Creating a query
- Manipulating a query
- Introspection on the GDC API fields
- Executing an API call to retrieve query results
- Raw data file downloads
- Summarizing and aggregating field values (faceting)
- Authentication
- BAM file slicing
Usage
There are two main classes of operations when working with the NCI GDC.
- Querying metadata and finding data files (e.g., finding all gene expression quantifications data files for all colon cancer patients).
- Transferring raw or processed data from the GDC to another computer (e.g., downloading raw or processed data)
Both classes of operation are reviewed in detail in the following sections.
Querying metadata
Vast amounts of metadata about cases (patients, basically), files, projects, and so-called annotations are available via the NCI GDC API. Typically, one will want to query metadata to either focus in on a set of files for download or transfer or to perform so-called aggregations (pivot-tables, facets, similar to the R table() functionality).
Querying metadata starts with creating a “blank” query. One will often then want to filter the query to limit results prior to retrieving results. The GenomicDataCommons package has helper functions for listing fields that are available for filtering.
In addition to fetching results, the GDC API allows faceting, or aggregating,, useful for compiling reports, generating dashboards, or building user interfaces to GDC data (see GDC web query interface for a non-R-based example).
Creating a query
The GenomicDataCommons package accesses the same API as the GDC website. Therefore, a useful approach, particularly for beginning users is to examine the filters available on the GDC repository pages to find appropriate filtering criteria. From there, converting those checkboxes to a GenomicDataCommons query() is relatively straightforward. Note that only a small subset of the available_fields() are available by default on the website.
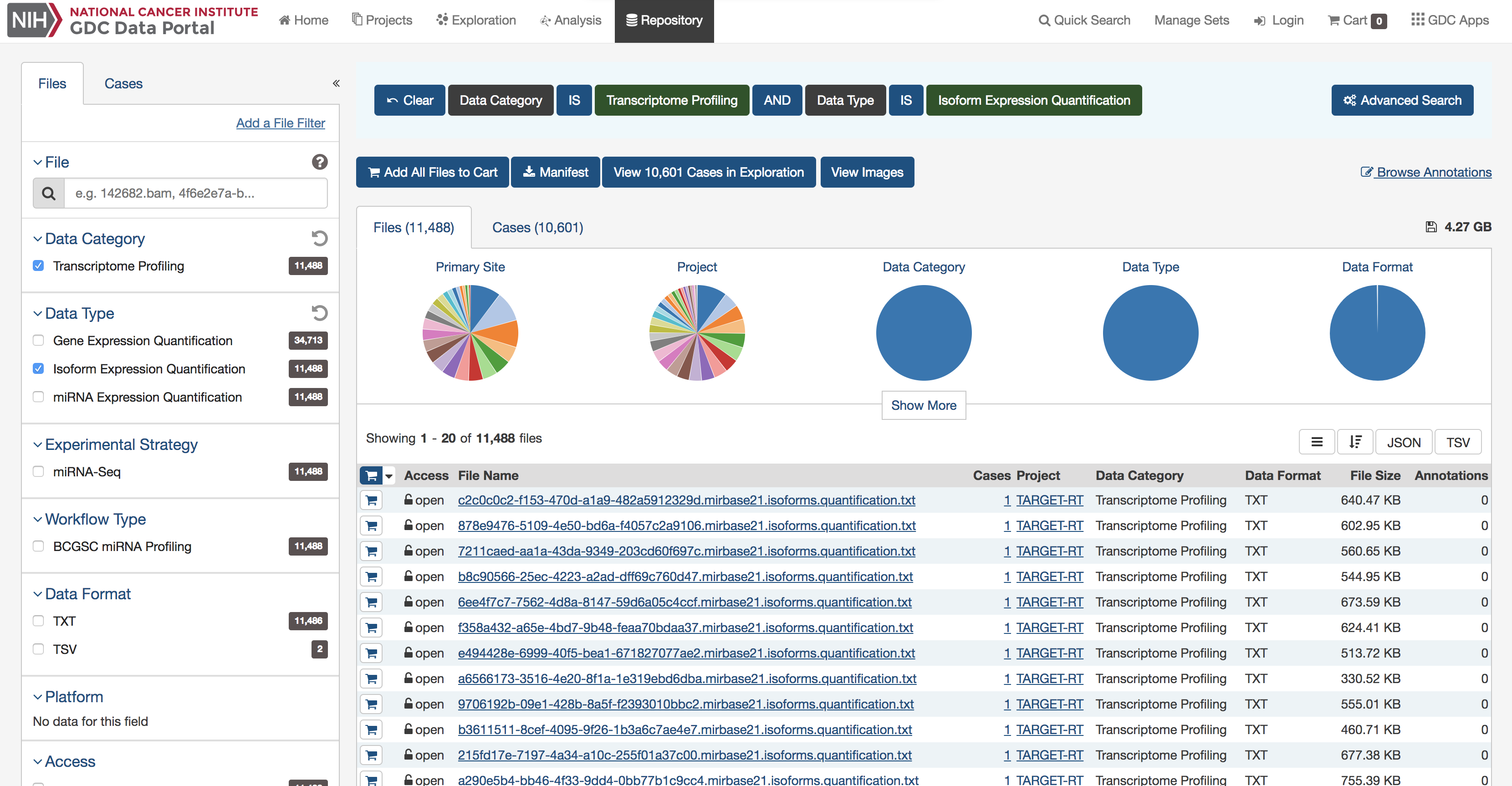
A screenshot of an example query of the GDC repository portal.
A query of the GDC starts its life in R. Queries follow the four metadata endpoints available at the GDC. In particular, there are four convenience functions that each create GDCQuery objects (actually, specific subclasses of GDCQuery):
pquery = projects()The pquery object is now an object of (S3) class, GDCQuery (and gdc_projects and list). The object contains the following elements:
- fields: This is a character vector of the fields that will be returned when we retrieve data. If no fields are specified to, for example, the
projects()function, the default fields from the GDC are used (seedefault_fields()) - filters: This will contain results after calling the
filter()method and will be used to filter results on retrieval. - facets: A character vector of field names that will be used for aggregating data in a call to
aggregations(). - archive: One of either “default” or “legacy”.
- token: A character(1) token from the GDC. See the authentication section for details, but note that, in general, the token is not necessary for metadata query and retrieval, only for actual data download.
Looking at the actual object (get used to using str()!), note that the query contains no results.
str(pquery)## List of 5
## $ fields : chr [1:10] "dbgap_accession_number" "disease_type" "intended_release_date" "name" ...
## $ filters: NULL
## $ facets : NULL
## $ legacy : logi FALSE
## $ expand : NULL
## - attr(*, "class")= chr [1:3] "gdc_projects" "GDCQuery" "list"Retrieving results
[ GDC pagination documentation ]
With a query object available, the next step is to retrieve results from the GDC. The GenomicDataCommons package. The most basic type of results we can get is a simple count() of records available that satisfy the filter criteria. Note that we have not set any filters, so a count() here will represent all the project records publicly available at the GDC in the “default” archive"
## [1] 68The results() method will fetch actual results.
presults = pquery %>% results()These results are returned from the GDC in JSON format and converted into a (potentially nested) list in R. The str() method is useful for taking a quick glimpse of the data.
str(presults)## List of 9
## $ id : chr [1:10] "GENIE-MSK" "TCGA-UCEC" "TCGA-LGG" "TCGA-SARC" ...
## $ primary_site :List of 10
## ..$ GENIE-MSK : chr [1:49] "Testis" "Gallbladder" "Unknown" "Other and unspecified parts of biliary tract" ...
## ..$ TCGA-UCEC : chr [1:2] "Corpus uteri" "Uterus, NOS"
## ..$ TCGA-LGG : chr "Brain"
## ..$ TCGA-SARC : chr [1:13] "Kidney" "Other and unspecified parts of tongue" "Bones, joints and articular cartilage of limbs" "Colon" ...
## ..$ TCGA-PAAD : chr "Pancreas"
## ..$ TCGA-ESCA : chr [1:2] "Esophagus" "Stomach"
## ..$ TCGA-PRAD : chr "Prostate gland"
## ..$ GENIE-VICC: chr [1:46] "Testis" "Unknown" "Other and unspecified parts of biliary tract" "Adrenal gland" ...
## ..$ TCGA-LAML : chr "Hematopoietic and reticuloendothelial systems"
## ..$ TCGA-KIRC : chr "Kidney"
## $ dbgap_accession_number: logi [1:10] NA NA NA NA NA NA ...
## $ project_id : chr [1:10] "GENIE-MSK" "TCGA-UCEC" "TCGA-LGG" "TCGA-SARC" ...
## $ disease_type :List of 10
## ..$ GENIE-MSK : chr [1:49] "Germ Cell Neoplasms" "Granular Cell Tumors and Alveolar Soft Part Sarcomas" "Immunoproliferative Diseases" "Plasma Cell Tumors" ...
## ..$ TCGA-UCEC : chr [1:4] "Epithelial Neoplasms, NOS" "Cystic, Mucinous and Serous Neoplasms" "Adenomas and Adenocarcinomas" "Not Reported"
## ..$ TCGA-LGG : chr "Gliomas"
## ..$ TCGA-SARC : chr [1:6] "Nerve Sheath Tumors" "Myomatous Neoplasms" "Fibromatous Neoplasms" "Lipomatous Neoplasms" ...
## ..$ TCGA-PAAD : chr [1:4] "Cystic, Mucinous and Serous Neoplasms" "Ductal and Lobular Neoplasms" "Adenomas and Adenocarcinomas" "Epithelial Neoplasms, NOS"
## ..$ TCGA-ESCA : chr [1:3] "Cystic, Mucinous and Serous Neoplasms" "Squamous Cell Neoplasms" "Adenomas and Adenocarcinomas"
## ..$ TCGA-PRAD : chr [1:3] "Cystic, Mucinous and Serous Neoplasms" "Ductal and Lobular Neoplasms" "Adenomas and Adenocarcinomas"
## ..$ GENIE-VICC: chr [1:41] "Germ Cell Neoplasms" "Acinar Cell Neoplasms" "Synovial-like Neoplasms" "Plasma Cell Tumors" ...
## ..$ TCGA-LAML : chr "Myeloid Leukemias"
## ..$ TCGA-KIRC : chr "Adenomas and Adenocarcinomas"
## $ name : chr [1:10] "AACR Project GENIE - Contributed by Memorial Sloan Kettering Cancer Center" "Uterine Corpus Endometrial Carcinoma" "Brain Lower Grade Glioma" "Sarcoma" ...
## $ releasable : logi [1:10] FALSE TRUE TRUE TRUE TRUE TRUE ...
## $ state : chr [1:10] "open" "open" "open" "open" ...
## $ released : logi [1:10] TRUE TRUE TRUE TRUE TRUE TRUE ...
## - attr(*, "row.names")= int [1:10] 1 2 3 4 5 6 7 8 9 10
## - attr(*, "class")= chr [1:3] "GDCprojectsResults" "GDCResults" "list"A default of only 10 records are returned. We can use the size and from arguments to results() to either page through results or to change the number of results. Finally, there is a convenience method, results_all() that will simply fetch all the available results given a query. Note that results_all() may take a long time and return HUGE result sets if not used carefully. Use of a combination of count() and results() to get a sense of the expected data size is probably warranted before calling results_all()
## [1] 10
presults = pquery %>% results_all()
length(ids(presults))## [1] 68## [1] TRUEExtracting subsets of results or manipulating the results into a more conventional R data structure is not easily generalizable. However, the purrr, rlist, and data.tree packages are all potentially of interest for manipulating complex, nested list structures. For viewing the results in an interactive viewer, consider the listviewer package.
Fields and Values
Central to querying and retrieving data from the GDC is the ability to specify which fields to return, filtering by fields and values, and faceting or aggregating. The GenomicDataCommons package includes two simple functions, available_fields() and default_fields(). Each can operate on a character(1) endpoint name (“cases,” “files,” “annotations,” or “projects”) or a GDCQuery object.
default_fields('files')## [1] "access" "acl"
## [3] "average_base_quality" "average_insert_size"
## [5] "average_read_length" "channel"
## [7] "chip_id" "chip_position"
## [9] "contamination" "contamination_error"
## [11] "created_datetime" "data_category"
## [13] "data_format" "data_type"
## [15] "error_type" "experimental_strategy"
## [17] "file_autocomplete" "file_id"
## [19] "file_name" "file_size"
## [21] "imaging_date" "magnification"
## [23] "md5sum" "mean_coverage"
## [25] "msi_score" "msi_status"
## [27] "pairs_on_diff_chr" "plate_name"
## [29] "plate_well" "platform"
## [31] "proportion_base_mismatch" "proportion_coverage_10x"
## [33] "proportion_coverage_10X" "proportion_coverage_30x"
## [35] "proportion_coverage_30X" "proportion_reads_duplicated"
## [37] "proportion_reads_mapped" "proportion_targets_no_coverage"
## [39] "read_pair_number" "revision"
## [41] "stain_type" "state"
## [43] "state_comment" "submitter_id"
## [45] "tags" "total_reads"
## [47] "tumor_ploidy" "tumor_purity"
## [49] "type" "updated_datetime"
# The number of fields available for files endpoint
length(available_fields('files'))## [1] 943
# The first few fields available for files endpoint
head(available_fields('files'))## [1] "access" "acl"
## [3] "analysis.analysis_id" "analysis.analysis_type"
## [5] "analysis.created_datetime" "analysis.input_files.access"The fields to be returned by a query can be specified following a similar paradigm to that of the dplyr package. The select() function is a verb that resets the fields slot of a GDCQuery; note that this is not quite analogous to the dplyr select() verb that limits from already-present fields. We completely replace the fields when using select() on a GDCQuery.
# Default fields here
qcases = cases()
qcases$fields## [1] "aliquot_ids" "analyte_ids"
## [3] "case_autocomplete" "case_id"
## [5] "consent_type" "created_datetime"
## [7] "days_to_consent" "days_to_lost_to_followup"
## [9] "diagnosis_ids" "disease_type"
## [11] "index_date" "lost_to_followup"
## [13] "portion_ids" "primary_site"
## [15] "sample_ids" "slide_ids"
## [17] "state" "submitter_aliquot_ids"
## [19] "submitter_analyte_ids" "submitter_diagnosis_ids"
## [21] "submitter_id" "submitter_portion_ids"
## [23] "submitter_sample_ids" "submitter_slide_ids"
## [25] "updated_datetime"
# set up query to use ALL available fields
# Note that checking of fields is done by select()
qcases = cases() %>% GenomicDataCommons::select(available_fields('cases'))
head(qcases$fields)## [1] "case_id" "aliquot_ids"
## [3] "analyte_ids" "annotations.annotation_id"
## [5] "annotations.case_id" "annotations.case_submitter_id"Finding fields of interest is such a common operation that the GenomicDataCommons includes the grep_fields() function and the field_picker() widget. See the appropriate help pages for details.
Facets and aggregation
The GDC API offers a feature known as aggregation or faceting. By specifying one or more fields (of appropriate type), the GDC can return to us a count of the number of records matching each potential value. This is similar to the R table method. Multiple fields can be returned at once, but the GDC API does not have a cross-tabulation feature; all aggregations are only on one field at a time. Results of aggregation() calls come back as a list of data.frames (actually, tibbles).
# total number of files of a specific type
res = files() %>% facet(c('type','data_type')) %>% aggregations()
res$type## doc_count key
## 1 151077 annotated_somatic_mutation
## 2 88599 simple_somatic_mutation
## 3 82177 aligned_reads
## 4 61693 gene_expression
## 5 58116 copy_number_segment
## 6 45419 copy_number_estimate
## 7 30072 slide_image
## 8 29990 mirna_expression
## 9 25591 biospecimen_supplement
## 10 12931 clinical_supplement
## 11 12359 methylation_beta_value
## 12 11444 structural_variation
## 13 4359 aggregated_somatic_mutation
## 14 4317 masked_somatic_mutation
## 15 54 secondary_expression_analysisUsing aggregations() is an also easy way to learn the contents of individual fields and forms the basis for faceted search pages.
Filtering
[ GDC filtering documentation ]
The GenomicDataCommons package uses a form of non-standard evaluation to specify R-like queries that are then translated into an R list. That R list is, upon calling a method that fetches results from the GDC API, translated into the appropriate JSON string. The R expression uses the formula interface as suggested by Hadley Wickham in his vignette on non-standard evaluation
It’s best to use a formula because a formula captures both the expression to evaluate and the environment where the evaluation occurs. This is important if the expression is a mixture of variables in a data frame and objects in the local environment [for example].
For the user, these details will not be too important except to note that a filter expression must begin with a “~.”
## [1] 618198To limit the file type, we can refer back to the section on faceting to see the possible values for the file field “type.” For example, to filter file results to only “gene_expression” files, we simply specify a filter.
qfiles = files() %>% filter(~ type == 'gene_expression')
# here is what the filter looks like after translation
str(get_filter(qfiles))## List of 2
## $ op : 'scalar' chr "="
## $ content:List of 2
## ..$ field: chr "type"
## ..$ value: chr "gene_expression"What if we want to create a filter based on the project (‘TCGA-OVCA,’ for example)? Well, we have a couple of possible ways to discover available fields. The first is based on base R functionality and some intuition.
grep('pro', available_fields('files'), value=TRUE)## [1] "analysis.input_files.proportion_base_mismatch"
## [2] "analysis.input_files.proportion_coverage_10x"
## [3] "analysis.input_files.proportion_coverage_10X"
## [4] "analysis.input_files.proportion_coverage_30x"
## [5] "analysis.input_files.proportion_coverage_30X"
## [6] "analysis.input_files.proportion_reads_duplicated"
## [7] "analysis.input_files.proportion_reads_mapped"
## [8] "analysis.input_files.proportion_targets_no_coverage"
## [9] "cases.diagnoses.international_prognostic_index"
## [10] "cases.diagnoses.progression_or_recurrence"
## [11] "cases.follow_ups.days_to_progression"
## [12] "cases.follow_ups.days_to_progression_free"
## [13] "cases.follow_ups.procedures_performed"
## [14] "cases.follow_ups.progression_or_recurrence"
## [15] "cases.follow_ups.progression_or_recurrence_anatomic_site"
## [16] "cases.follow_ups.progression_or_recurrence_type"
## [17] "cases.project.dbgap_accession_number"
## [18] "cases.project.disease_type"
## [19] "cases.project.intended_release_date"
## [20] "cases.project.name"
## [21] "cases.project.primary_site"
## [22] "cases.project.program.dbgap_accession_number"
## [23] "cases.project.program.name"
## [24] "cases.project.program.program_id"
## [25] "cases.project.project_id"
## [26] "cases.project.releasable"
## [27] "cases.project.released"
## [28] "cases.project.state"
## [29] "cases.samples.days_to_sample_procurement"
## [30] "cases.samples.method_of_sample_procurement"
## [31] "cases.samples.portions.slides.number_proliferating_cells"
## [32] "cases.samples.portions.slides.prostatic_chips_positive_count"
## [33] "cases.samples.portions.slides.prostatic_chips_total_count"
## [34] "cases.samples.portions.slides.prostatic_involvement_percent"
## [35] "cases.tissue_source_site.project"
## [36] "downstream_analyses.output_files.proportion_base_mismatch"
## [37] "downstream_analyses.output_files.proportion_coverage_10x"
## [38] "downstream_analyses.output_files.proportion_coverage_10X"
## [39] "downstream_analyses.output_files.proportion_coverage_30x"
## [40] "downstream_analyses.output_files.proportion_coverage_30X"
## [41] "downstream_analyses.output_files.proportion_reads_duplicated"
## [42] "downstream_analyses.output_files.proportion_reads_mapped"
## [43] "downstream_analyses.output_files.proportion_targets_no_coverage"
## [44] "index_files.proportion_base_mismatch"
## [45] "index_files.proportion_coverage_10x"
## [46] "index_files.proportion_coverage_10X"
## [47] "index_files.proportion_coverage_30x"
## [48] "index_files.proportion_coverage_30X"
## [49] "index_files.proportion_reads_duplicated"
## [50] "index_files.proportion_reads_mapped"
## [51] "index_files.proportion_targets_no_coverage"
## [52] "proportion_base_mismatch"
## [53] "proportion_coverage_10x"
## [54] "proportion_coverage_10X"
## [55] "proportion_coverage_30x"
## [56] "proportion_coverage_30X"
## [57] "proportion_reads_duplicated"
## [58] "proportion_reads_mapped"
## [59] "proportion_targets_no_coverage"Interestingly, the project information is “nested” inside the case. We don’t need to know that detail other than to know that we now have a few potential guesses for where our information might be in the files records. We need to know where because we need to construct the appropriate filter.
files() %>% facet('cases.project.project_id') %>% aggregations()## $cases.project.project_id
## doc_count key
## 1 36134 FM-AD
## 2 33766 TCGA-BRCA
## 3 42178 CPTAC-3
## 4 36470 GENIE-MSK
## 5 18358 TCGA-LUAD
## 6 17277 TCGA-UCEC
## 7 16340 TCGA-HNSC
## 8 16344 TCGA-OV
## 9 15445 TCGA-THCA
## 10 29433 MMRF-COMMPASS
## 11 16368 TCGA-LUSC
## 12 15795 TCGA-LGG
## 13 28464 GENIE-DFCI
## 14 16255 TCGA-KIRC
## 15 15296 TCGA-PRAD
## 16 15338 TCGA-COAD
## 17 13089 TCGA-GBM
## 18 20772 TARGET-ALL-P2
## 19 13674 TCGA-SKCM
## 20 13739 TCGA-STAD
## 21 12513 TCGA-BLCA
## 22 11578 TCGA-LIHC
## 23 9201 TCGA-CESC
## 24 9137 TCGA-KIRP
## 25 8002 TCGA-SARC
## 26 7772 TARGET-AML
## 27 5671 TCGA-PAAD
## 28 5657 TCGA-ESCA
## 29 5378 TCGA-PCPG
## 30 5269 TCGA-READ
## 31 5796 TARGET-NBL
## 32 8981 BEATAML1.0-COHORT
## 33 8958 CPTAC-2
## 34 4605 TCGA-TGCT
## 35 4814 TCGA-LAML
## 36 3691 TCGA-THYM
## 37 6036 HCMI-CMDC
## 38 5941 CMI-MBC
## 39 5550 CGCI-HTMCP-CC
## 40 2736 TCGA-ACC
## 41 2457 TCGA-KICH
## 42 2677 TARGET-WT
## 43 4805 NCICCR-DLBCL
## 44 2518 TCGA-MESO
## 45 2340 TCGA-UVM
## 46 3113 TARGET-OS
## 47 3982 TARGET-ALL-P3
## 48 3857 GENIE-MDA
## 49 3833 GENIE-VICC
## 50 3320 GENIE-JHU
## 51 1765 TCGA-UCS
## 52 1426 TCGA-CHOL
## 53 2632 GENIE-UHN
## 54 1325 TCGA-DLBC
## 55 2477 CGCI-BLGSP
## 56 1049 TARGET-RT
## 57 1038 GENIE-GRCC
## 58 994 WCDT-MCRPC
## 59 934 CMI-ASC
## 60 801 GENIE-NKI
## 61 798 OHSU-CNL
## 62 703 ORGANOID-PANCREATIC
## 63 570 CMI-MPC
## 64 417 CTSP-DLBCL1
## 65 223 BEATAML1.0-CRENOLANIB
## 66 169 TARGET-CCSK
## 67 133 TARGET-ALL-P1
## 68 21 VAREPOP-APOLLOWe note that cases.project.project_id looks like it is a good fit. We also note that TCGA-OV is the correct project_id, not TCGA-OVCA. Note that unlike with dplyr and friends, the filter() method here replaces the filter and does not build on any previous filters.
qfiles = files() %>%
filter( ~ cases.project.project_id == 'TCGA-OV' & type == 'gene_expression')
str(get_filter(qfiles))## List of 2
## $ op : 'scalar' chr "and"
## $ content:List of 2
## ..$ :List of 2
## .. ..$ op : 'scalar' chr "="
## .. ..$ content:List of 2
## .. .. ..$ field: chr "cases.project.project_id"
## .. .. ..$ value: chr "TCGA-OV"
## ..$ :List of 2
## .. ..$ op : 'scalar' chr "="
## .. ..$ content:List of 2
## .. .. ..$ field: chr "type"
## .. .. ..$ value: chr "gene_expression"
qfiles %>% count()## [1] 1137Asking for a count() of results given these new filter criteria gives r qfiles %>% count() results. Generating a manifest for bulk downloads is as simple as asking for the manifest from the current query.
## # A tibble: 6 x 5
## id filename md5 size state
## <chr> <chr> <chr> <dbl> <chr>
## 1 4b25720c-2fad-4055… 51b8dd7e-1686-46bb-9a29-b0… 19af96c6c5d4e11… 538565 relea…
## 2 5e8d1839-40ab-432b… 9f223240-1020-4450-a6ec-a6… 968c18169c965ee… 568707 relea…
## 3 691ec8c8-17b3-4c26… 0315439e-694c-4b14-a27c-29… 2cd3130425a20de… 252344 relea…
## 4 18863638-3369-4ca3… 4d337bb6-8e53-4142-8c22-43… bda0aeee9382d72… 249487 relea…
## 5 81c89a1f-19d6-43df… ce511378-d8f8-494e-a07d-2e… 6d82eb3ca6b62b2… 559993 relea…
## 6 fe36a6b6-6401-4fc5… 968544ba-7990-41e8-a0ee-52… b382541b102c7e1… 526190 relea…Note that we might still not be quite there. Looking at filenames, there are suspiciously named files that might include “FPKM,” “FPKM-UQ,” or “counts.” Another round of grep and available_fields, looking for “type” turned up that the field “analysis.workflow_type” has the appropriate filter criteria.
qfiles = files() %>% filter( ~ cases.project.project_id == 'TCGA-OV' &
type == 'gene_expression' &
analysis.workflow_type == 'HTSeq - Counts')
manifest_df = qfiles %>% manifest()
nrow(manifest_df)## [1] 379The GDC Data Transfer Tool can be used (from R, transfer() or from the command-line) to orchestrate high-performance, restartable transfers of all the files in the manifest. See the bulk downloads section for details.
Authentication
[ GDC authentication documentation ]
The GDC offers both “controlled-access” and “open” data. As of this writing, only data stored as files is “controlled-access”; that is, metadata accessible via the GDC is all “open” data and some files are “open” and some are “controlled-access.” Controlled-access data are only available after going through the process of obtaining access.
After controlled-access to one or more datasets has been granted, logging into the GDC web portal will allow you to access a GDC authentication token, which can be downloaded and then used to access available controlled-access data via the GenomicDataCommons package.
The GenomicDataCommons uses authentication tokens only for downloading data (see transfer and gdcdata documentation). The package includes a helper function, gdc_token, that looks for the token to be stored in one of three ways (resolved in this order):
- As a string stored in the environment variable,
GDC_TOKEN - As a file, stored in the file named by the environment variable,
GDC_TOKEN_FILE - In a file in the user home directory, called
.gdc_token
As a concrete example:
Datafile access and download
The gdcdata function takes a character vector of one or more file ids. A simple way of producing such a vector is to produce a manifest data frame and then pass in the first column, which will contain file ids.
fnames = gdcdata(manifest_df$id[1:2],progress=FALSE)Note that for controlled-access data, a GDC authentication token is required. Using the BiocParallel package may be useful for downloading in parallel, particularly for large numbers of smallish files.
The bulk download functionality is only efficient (as of v1.2.0 of the GDC Data Transfer Tool) for relatively large files, so use this approach only when transferring BAM files or larger VCF files, for example. Otherwise, consider using the approach shown above, perhaps in parallel.
fnames = gdcdata(manifest_df$id[3:10], access_method = 'client')Benjamin Haibe-Kains
Pharmacogenomics
Pharmacogenomics holds great promise for the development of biomarkers of drug response and the design of new therapeutic options, which are key challenges in precision medicine. However, such data are scattered and lack standards for efficient access and analysis, consequently preventing the realization of the full potential of pharmacogenomics. To address these issues, we implemented PharmacoGx, an easy-to-use, open source package for integrative analysis of multiple pharmacogenomic datasets. PharmacoGx provides a unified framework for downloading and analyzing large pharmacogenomic datasets which are extensively curated to ensure maximum overlap and consistency.
Examples of PharmacoGx usage in biomedical research can be found in the following publications:
- Smirnov et al. PharmacoGx: an R package for analysis of large pharmacogenomic datasets." Bioinformatics (2015): 1244-1246.
- Yao et al., Tissue specificity of in vitro drug sensitivity, JAMIA (2017)
- Safikhani et al., Gene isoforms as expression-based biomarkers predictive of drug response in vitro, Nature Communications (2017)
- El-Hachem et al., Integrative Cancer Pharmacogenomics to Infer Large-Scale Drug Taxonomy, Cancer Research (2017)
- Singh, M. et al. Therapeutic Targeting of the Premetastatic Stage in Human Lung-to-Brain Metastasis. Cancer Research (2018).
- Knowles, D. A., Bouchard, G. & Plevritis, S. Sparse discriminative latent characteristics for predicting cancer drug sensitivity from genomic features. PLOS Computational Biology (2019)
- Corsello, S. M. et al. Discovering the anticancer potential of non-oncology drugs by systematic viability profiling, Nat Cancer (2020)
Getting started
Let us first load the PharmacoGx library.
library(PharmacoGx)## Warning: no DISPLAY variable so Tk is not availableWe can now access large-scale preclinical pharmacogenomic datasets that have been fully curated for ease of use.
Overview of PharmacoGx datasets (PharmacoSets)
To efficiently store and analyze large pharmacogenomic datasets, we developed the PharmacoSet class (also referred to as PSet), which acts as a data container storing pharmacological and molecular data along with experimental metadata (detailed structure provided in Supplementary materials). This class enables efficient implementation of curated annotations for cell lines, drug compounds and molecular features, which facilitates comparisons between different datasets stored as PharmacoSet objects.
We have made the PharmacoSet objects of the curated datasets available for download using functions provided in the package. A table of available PharmacoSet objects can be obtained by using the availablePSets function. Any of the PharmacoSets in the table can then be downloaded by calling downloadPSet, which saves the datasets into a directory of the users choice, and returns the data into the R session.
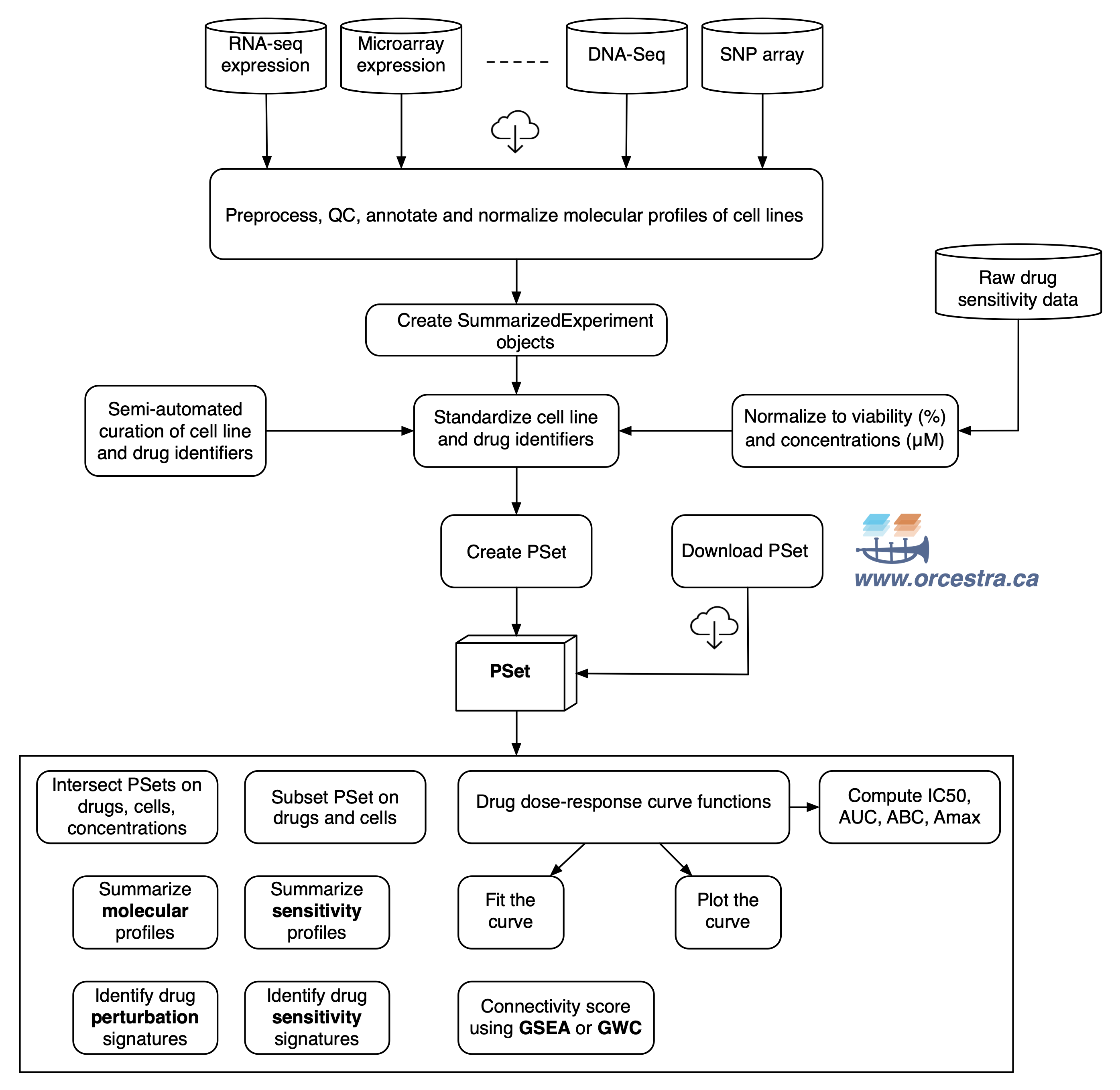
Overview of PharmacoGx
To get a list of all the available PharmacoSets in PharmacoGx, we can use the availablePSets` function, which returns a table providing key information for each dataset.
(psets <- PharmacoGx::availablePSets()[,-c(6,7)])## Dataset Name Date Created PSet Name version
## 1 CCLE 2020-06-24T14:39:26.588Z CCLE_2015 2015
## 2 CTRPv2 2020-06-24T14:39:26.588Z CTRPv2_2015 2015
## 3 FIMM 2020-06-24T14:39:26.588Z FIMM_2016 2016
## 4 gCSI 2020-06-24T14:39:26.588Z gCSI_2017 2017
## 5 GDSC 2020-06-24T14:39:26.588Z GDSC_2020(v2-8.2) 2020(v2-8.2)
## 6 GDSC 2020-06-24T14:39:26.588Z GDSC_2020(v1-8.2) 2020(v1-8.2)
## 7 GRAY 2021-02-23T14:39:26.588Z GRAY_2017 2017
## 8 UHNBreast 2020-06-24T14:39:26.588Z UHNBreast_2019 2019
## type DOI
## 1 sensitivity 10.5281/zenodo.3905462
## 2 sensitivity 10.5281/zenodo.3905470
## 3 sensitivity 10.5281/zenodo.3905448
## 4 sensitivity 10.5281/zenodo.3905452
## 5 sensitivity 10.5281/zenodo.3905481
## 6 sensitivity 10.5281/zenodo.3905485
## 7 sensitivity 10.5281/zenodo.4557735
## 8 both 10.5281/zenodo.3905460
## Download
## 1 https://zenodo.org/record/3905462/files/CCLE.rds?download=1
## 2 https://zenodo.org/record/3905470/files/CTRPv2.rds?download=1
## 3 https://zenodo.org/record/3905448/files/FIMM.rds?download=1
## 4 https://zenodo.org/record/3905452/files/gCSI.rds?download=1
## 5 https://zenodo.org/record/3905481/files/GDSC2.rds?download=1
## 6 https://zenodo.org/record/3905485/files/GDSC1.rds?download=1
## 7 https://zenodo.org/record/4557735/files/GRAY2017.rds?download=1
## 8 https://zenodo.org/record/3905460/files/UHNBreast.rds?download=1There are currently 8 datasets available in PharmacoGx, including sensitivity datasets and perturbation datasets (see below).
Drug Sensitivity Datasets
Drug sensitivity datasets refer to pharmacogenomic data where cancer cells are molecularly profiled at baseline (before drug treatment), and the effect of drug treatment on cell viability is measured using a pharmacological assay (e.g., Cell Titer-Glo). These datasets can be used for biomarker discovery by correlating the molecular features of cancer cells to their response to drugs of interest.
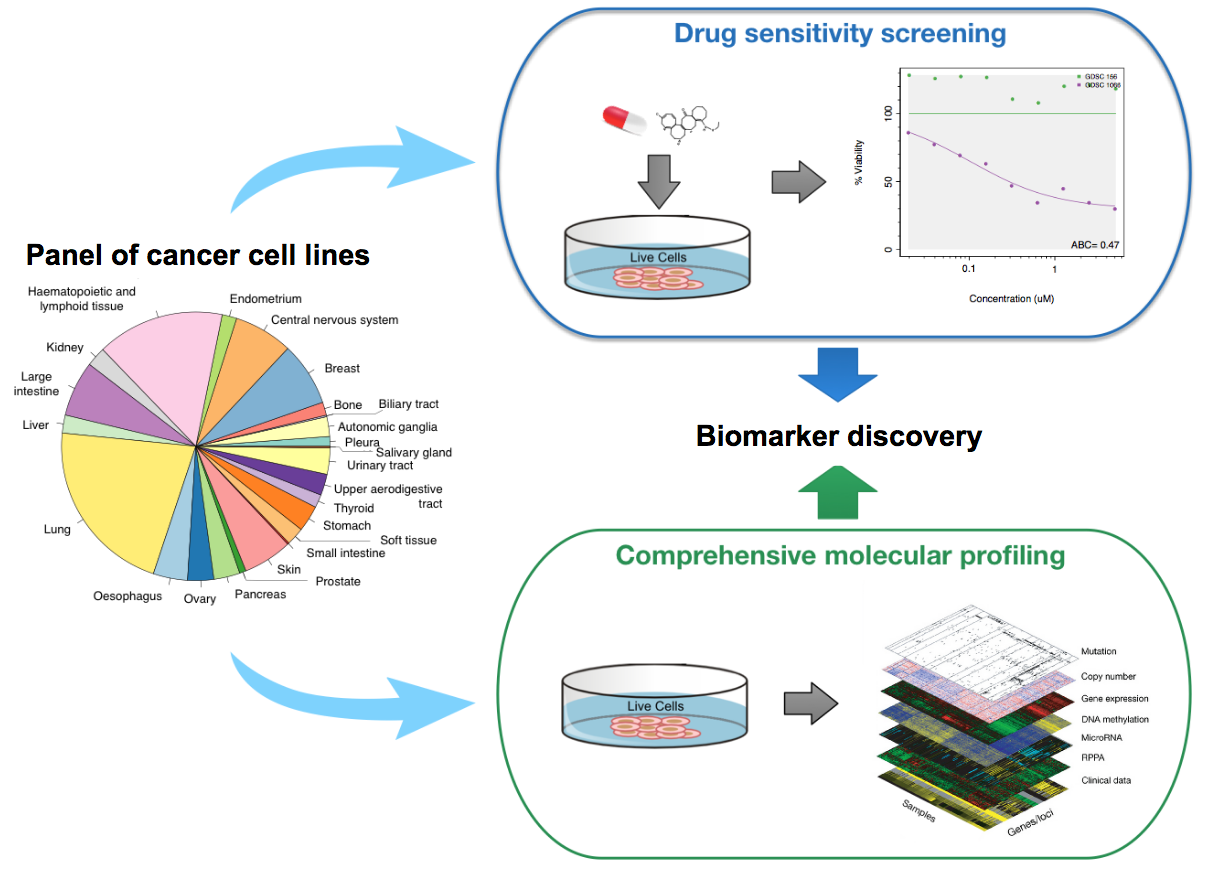
Schematic view of the drug sensitivity datasets.
Notably, the Genomics of Drug Sensitivity in Cancer GDSC and the Cancer Cell Line Encyclopedia CCLE are large drug sensitivity datasets published in seminal studies in Nature, Garnett et al., https://www.nature.com/articles/nature11005, Nature (2012) and Barretina et al., The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity, Nature (2012), respectively.
Drug Perturbation Datasets
Drug perturbation datasets refer to pharmacogenomic data where gene expression profiles are measured before and after short-term or medium term (e.g., 6h, 24h) drug treatment to identify genes that are up- and down-regulated due to the drug treatment. These datasets can be to classify drug (drug taxonomy), infer their mechanism of action, or find drugs with similar effects (drug repurposing).
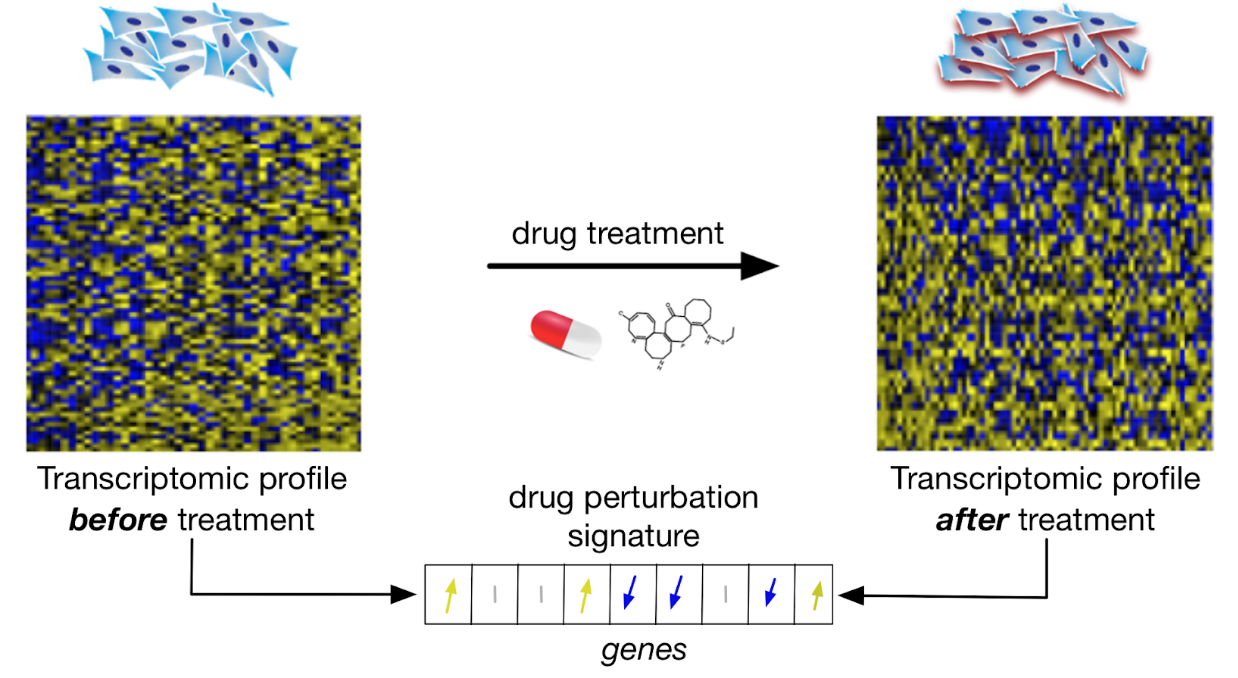
Schematic view of drug perturbation datasets
Large drug perturbation data have been generated within the Connectivity Map Project CAMP, with CMAPv2 and CMAPv3 available from PharmacoGx, published in Lamb et al., The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease, Science (2006) and Subramanian et al., A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles, Cell (2017), respectively.
Exploring Other Treatment Types
In addition to PharmacoGx, there is a suite of packages in Bioconductor for exploring public high throughput screening data. For Sensitivity datasets, Xeva provides access to public drug screening datasets in Patient Derived Xenograft models, including providing access to the Novartis PDX Encyclopedia dataset, published in Gao, H. et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response, Nature Medicine.. Additionally, RadioGx is currently available in the development branch of Bioconductor, providing access to cell line screening data for response to radiation treatment, featuring data from Yard, B. D. et al. A genetic basis for the variation in the vulnerability of cancer to DNA damage. Nature Communications 7, 11428 (2016)..
The Biomarker Discovery from High Throughput Screening Datasets workshop at Bioc2020 goes into depth about using these packages for accessing and exploring public sensitivity datasets.
In addition, Bioconductor also includes the ToxicoGx package, which provides access to Sensitivity and Perturbation datasets characterizing in vitro response of human tissue to toxicant exposure, currently providing access to data from the TGGates and DrugMatrix datasets. More information about the ToxicoGx package can be found at the Bioc2020 poster presentation on the package.
Levi Waldron
Accessing The Cancer Genome Atlas (TCGA)
We summarize two approaches to accessing TCGA data:
-
TCGAbiolinks:
- data access through GenomicDataCommons
- provides data both from the legacy Firehose pipeline used by the TCGA publications (alignments based on hg18 and hg19 builds2), and the GDC harmonized GRCh38 pipeline3.
- downloads files from the Genomic Data Commons, and provides conversion to
(Ranged)SummarizedExperimentwhere possible
-
curatedTCGAData:
- data access through ExperimentHub
- provides data from the legacy Firehose pipeline4
- provides individual assays as
(Ranged)SummarizedExperimentandRaggedExperiment, integrates multiple assays within and across cancer types using MultiAssayExperiment
TCGAbiolinks
We demonstrate here generating a RangedSummarizedExperiment for RNA-seq data from adrenocortical carcinoma (ACC). For additional information and options, see the TCGAbiolinks vignettes5.
Load packages:
Search for matching data:
library(TCGAbiolinks)
library(SummarizedExperiment)
query <- GDCquery(project = "TCGA-ACC",
data.category = "Gene expression",
data.type = "Gene expression quantification",
platform = "Illumina HiSeq",
file.type = "normalized_results",
experimental.strategy = "RNA-Seq",
legacy = TRUE)Download data and convert it to RangedSummarizedExperiment:
#gdcdir <- file.path("Waldron_PublicData", "GDCdata")
#GDCdownload(query, method = "api", files.per.chunk = 10,
# directory = gdcdir)
#ACCse <- GDCprepare(query, directory = gdcdir)
#ACCsecuratedTCGAData: Curated Data From The Cancer Genome Atlas as MultiAssayExperiment Objects
curatedTCGAData does not interface with the Genomic Data Commons, but downloads data from Bioconductor’s ExperimentHub.
References: * https://waldronlab.io/MultiAssayWorkshop/articles/curatedTCGAData_ref.html : available datasets and data types. * https://waldronlab.io/MultiAssayWorkshop/articles/TCGAutilsCheatsheet.html : quick-reference for related utilities in TCGAutils package * https://waldronlab.io/MultiAssayWorkshop/articles/Ramos_MultiAssayExperiment.html: workshop for MultiAssayExperiment
library(curatedTCGAData)
library(MultiAssayExperiment)By default, the curatedTCGAData() function will only show available datasets, and not download anything. The arguments are shown here only for demonstration, the same result is obtained with no arguments:
curatedTCGAData(diseaseCode = "*", assays = "*")## using temporary cache /tmp/RtmpkiIQ1v/BiocFileCache## snapshotDate(): 2020-10-27## See '?curatedTCGAData' for 'diseaseCode' and 'assays' inputs## ah_id title file_size
## 1 EH558 ACC_CNASNP-20160128 0.8 Mb
## 2 EH559 ACC_CNVSNP-20160128 0.2 Mb
## 3 EH561 ACC_GISTIC_AllByGene-20160128 0.3 Mb
## 4 EH2115 ACC_GISTIC_Peaks-20160128 0 Mb
## 5 EH562 ACC_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 6 EH2116 ACC_Methylation-20160128_assays 236.4 Mb
## 7 EH2117 ACC_Methylation-20160128_se 6.1 Mb
## 8 EH565 ACC_miRNASeqGene-20160128 0.1 Mb
## 9 EH566 ACC_Mutation-20160128 0.7 Mb
## 10 EH567 ACC_RNASeq2GeneNorm-20160128 4 Mb
## 11 EH568 ACC_RPPAArray-20160128 0.1 Mb
## 12 EH570 BLCA_CNASeq-20160128 0.3 Mb
## 13 EH571 BLCA_CNASNP-20160128 4.3 Mb
## 14 EH572 BLCA_CNVSNP-20160128 1.1 Mb
## 15 EH574 BLCA_GISTIC_AllByGene-20160128 0.6 Mb
## 16 EH2118 BLCA_GISTIC_Peaks-20160128 0 Mb
## 17 EH575 BLCA_GISTIC_ThresholdedByGene-20160128 0.3 Mb
## 18 EH2119 BLCA_Methylation-20160128_assays 1283.4 Mb
## 19 EH2120 BLCA_Methylation-20160128_se 6.1 Mb
## 20 EH578 BLCA_miRNASeqGene-20160128 0.4 Mb
## 21 EH579 BLCA_Mutation-20160128 3.7 Mb
## 22 EH580 BLCA_RNASeq2GeneNorm-20160128 21.9 Mb
## 23 EH581 BLCA_RNASeqGene-20160128 2.4 Mb
## 24 EH582 BLCA_RPPAArray-20160128 0.5 Mb
## 25 EH584 BRCA_CNASeq-20160128 0 Mb
## 26 EH585 BRCA_CNASNP-20160128 9.8 Mb
## 27 EH586 BRCA_CNVSNP-20160128 2.8 Mb
## 28 EH588 BRCA_GISTIC_AllByGene-20160128 1.3 Mb
## 29 EH2121 BRCA_GISTIC_Peaks-20160128 0 Mb
## 30 EH589 BRCA_GISTIC_ThresholdedByGene-20160128 0.4 Mb
## 31 EH2122 BRCA_Methylation_methyl27-20160128_assays 63.2 Mb
## 32 EH2123 BRCA_Methylation_methyl27-20160128_se 0.4 Mb
## 33 EH2124 BRCA_Methylation_methyl450-20160128_assays 2613.2 Mb
## 34 EH2125 BRCA_Methylation_methyl450-20160128_se 6.1 Mb
## 35 EH593 BRCA_miRNASeqGene-20160128 0.6 Mb
## 36 EH594 BRCA_mRNAArray-20160128 27.3 Mb
## 37 EH595 BRCA_Mutation-20160128 4.5 Mb
## 38 EH596 BRCA_RNASeq2GeneNorm-20160128 64.5 Mb
## 39 EH597 BRCA_RNASeqGene-20160128 30 Mb
## 40 EH598 BRCA_RPPAArray-20160128 1.6 Mb
## 41 EH600 CESC_CNASeq-20160128 0.1 Mb
## 42 EH601 CESC_CNASNP-20160128 2.3 Mb
## 43 EH602 CESC_CNVSNP-20160128 0.6 Mb
## 44 EH604 CESC_GISTIC_AllByGene-20160128 0.5 Mb
## 45 EH2126 CESC_GISTIC_Peaks-20160128 0 Mb
## 46 EH605 CESC_GISTIC_ThresholdedByGene-20160128 0.3 Mb
## 47 EH2127 CESC_Methylation-20160128_assays 921.2 Mb
## 48 EH2128 CESC_Methylation-20160128_se 6.1 Mb
## 49 EH608 CESC_miRNASeqGene-20160128 0.3 Mb
## 50 EH609 CESC_Mutation-20160128 2 Mb
## 51 EH610 CESC_RNASeq2GeneNorm-20160128 16.1 Mb
## 52 EH611 CESC_RPPAArray-20160128 0.3 Mb
## 53 EH613 CHOL_CNASNP-20160128 0.4 Mb
## 54 EH614 CHOL_CNVSNP-20160128 0.1 Mb
## 55 EH616 CHOL_GISTIC_AllByGene-20160128 0.3 Mb
## 56 EH2129 CHOL_GISTIC_Peaks-20160128 0 Mb
## 57 EH617 CHOL_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 58 EH2130 CHOL_Methylation-20160128_assays 132.1 Mb
## 59 EH2131 CHOL_Methylation-20160128_se 6.1 Mb
## 60 EH620 CHOL_miRNASeqGene-20160128 0.1 Mb
## 61 EH621 CHOL_Mutation-20160128 0.2 Mb
## 62 EH622 CHOL_RNASeq2GeneNorm-20160128 2.4 Mb
## 63 EH623 CHOL_RPPAArray-20160128 0.1 Mb
## 64 EH625 COAD_CNASeq-20160128 0.3 Mb
## 65 EH626 COAD_CNASNP-20160128 3.9 Mb
## 66 EH627 COAD_CNVSNP-20160128 0.9 Mb
## 67 EH629 COAD_GISTIC_AllByGene-20160128 0.5 Mb
## 68 EH2132 COAD_GISTIC_Peaks-20160128 0 Mb
## 69 EH630 COAD_GISTIC_ThresholdedByGene-20160128 0.3 Mb
## 70 EH2133 COAD_Methylation_methyl27-20160128_assays 37.2 Mb
## 71 EH2134 COAD_Methylation_methyl27-20160128_se 0.4 Mb
## 72 EH2135 COAD_Methylation_methyl450-20160128_assays 983.8 Mb
## 73 EH2136 COAD_Methylation_methyl450-20160128_se 6.1 Mb
## 74 EH634 COAD_miRNASeqGene-20160128 0.2 Mb
## 75 EH635 COAD_mRNAArray-20160128 8.1 Mb
## 76 EH636 COAD_Mutation-20160128 1.2 Mb
## 77 EH637 COAD_RNASeq2GeneNorm-20160128 8.8 Mb
## 78 EH638 COAD_RNASeqGene-20160128 0.4 Mb
## 79 EH639 COAD_RPPAArray-20160128 0.6 Mb
## 80 EH641 DLBC_CNASNP-20160128 0.4 Mb
## 81 EH642 DLBC_CNVSNP-20160128 0.1 Mb
## 82 EH644 DLBC_GISTIC_AllByGene-20160128 0.2 Mb
## 83 EH2137 DLBC_GISTIC_Peaks-20160128 0 Mb
## 84 EH645 DLBC_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 85 EH2138 DLBC_Methylation-20160128_assays 141.8 Mb
## 86 EH2139 DLBC_Methylation-20160128_se 6.1 Mb
## 87 EH648 DLBC_miRNASeqGene-20160128 0.1 Mb
## 88 EH649 DLBC_Mutation-20160128 0.7 Mb
## 89 EH650 DLBC_RNASeq2GeneNorm-20160128 2.5 Mb
## 90 EH651 DLBC_RPPAArray-20160128 0.1 Mb
## 91 EH653 ESCA_CNASeq-20160128 0.2 Mb
## 92 EH654 ESCA_CNASNP-20160128 1.8 Mb
## 93 EH655 ESCA_CNVSNP-20160128 0.7 Mb
## 94 EH657 ESCA_GISTIC_AllByGene-20160128 0.5 Mb
## 95 EH2140 ESCA_GISTIC_Peaks-20160128 0 Mb
## 96 EH658 ESCA_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 97 EH2141 ESCA_Methylation-20160128_assays 596.9 Mb
## 98 EH2142 ESCA_Methylation-20160128_se 6.1 Mb
## 99 EH661 ESCA_miRNASeqGene-20160128 0.2 Mb
## 100 EH662 ESCA_Mutation-20160128 2.8 Mb
## 101 EH663 ESCA_RNASeq2GeneNorm-20160128 10.8 Mb
## 102 EH664 ESCA_RNASeqGene-20160128 8 Mb
## 103 EH665 ESCA_RPPAArray-20160128 0.2 Mb
## 104 EH667 GBM_CNACGH_CGH_hg_244a-20160128 0.7 Mb
## 105 EH668 GBM_CNACGH_CGH_hg_415k_g4124a-20160128 0.5 Mb
## 106 EH669 GBM_CNASNP-20160128 5.2 Mb
## 107 EH670 GBM_CNVSNP-20160128 1.5 Mb
## 108 EH672 GBM_GISTIC_AllByGene-20160128 0.7 Mb
## 109 EH2143 GBM_GISTIC_Peaks-20160128 0 Mb
## 110 EH673 GBM_GISTIC_ThresholdedByGene-20160128 0.3 Mb
## 111 EH2144 GBM_Methylation_methyl27-20160128_assays 52.4 Mb
## 112 EH2145 GBM_Methylation_methyl27-20160128_se 0.4 Mb
## 113 EH2146 GBM_Methylation_methyl450-20160128_assays 455.1 Mb
## 114 EH2147 GBM_Methylation_methyl450-20160128_se 6.1 Mb
## 115 EH677 GBM_miRNAArray-20160128 2.1 Mb
## 116 EH678 GBM_miRNASeqGene-20160128 0 Mb
## 117 EH679 GBM_mRNAArray_huex-20160128 56.3 Mb
## 118 EH2148 GBM_mRNAArray_TX_g4502a_1-20160128 22.5 Mb
## 119 EH680 GBM_mRNAArray_TX_g4502a-20160128 5.7 Mb
## 120 EH681 GBM_mRNAArray_TX_ht_hg_u133a-20160128 44.7 Mb
## 121 EH682 GBM_Mutation-20160128 2.1 Mb
## 122 EH683 GBM_RNASeq2GeneNorm-20160128 8.6 Mb
## 123 EH684 GBM_RPPAArray-20160128 0.4 Mb
## 124 EH686 HNSC_CNASeq-20160128 0.3 Mb
## 125 EH687 HNSC_CNASNP-20160128 4.2 Mb
## 126 EH688 HNSC_CNVSNP-20160128 1.1 Mb
## 127 EH690 HNSC_GISTIC_AllByGene-20160128 0.6 Mb
## 128 EH2149 HNSC_GISTIC_Peaks-20160128 0 Mb
## 129 EH691 HNSC_GISTIC_ThresholdedByGene-20160128 0.3 Mb
## 130 EH2150 HNSC_Methylation-20160128_assays 1714.7 Mb
## 131 EH2151 HNSC_Methylation-20160128_se 6.1 Mb
## 132 EH694 HNSC_miRNASeqGene-20160128 0.4 Mb
## 133 EH695 HNSC_Mutation-20160128 4.8 Mb
## 134 EH696 HNSC_RNASeq2GeneNorm-20160128 29.5 Mb
## 135 EH697 HNSC_RNASeqGene-20160128 10.3 Mb
## 136 EH698 HNSC_RPPAArray-20160128 0.3 Mb
## 137 EH700 KICH_CNASNP-20160128 0.5 Mb
## 138 EH701 KICH_CNVSNP-20160128 0.1 Mb
## 139 EH703 KICH_GISTIC_AllByGene-20160128 0.2 Mb
## 140 EH2152 KICH_GISTIC_Peaks-20160128 0 Mb
## 141 EH704 KICH_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 142 EH2153 KICH_Methylation-20160128_assays 195 Mb
## 143 EH2154 KICH_Methylation-20160128_se 6.1 Mb
## 144 EH707 KICH_miRNASeqGene-20160128 0.1 Mb
## 145 EH708 KICH_Mutation-20160128 0.1 Mb
## 146 EH709 KICH_RNASeq2GeneNorm-20160128 4.9 Mb
## 147 EH710 KICH_RPPAArray-20160128 0.1 Mb
## 148 EH712 KIRC_CNASNP-20160128 4.1 Mb
## 149 EH713 KIRC_CNVSNP-20160128 0.8 Mb
## 150 EH715 KIRC_GISTIC_AllByGene-20160128 0.5 Mb
## 151 EH2155 KIRC_GISTIC_Peaks-20160128 0 Mb
## 152 EH716 KIRC_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 153 EH2156 KIRC_Methylation_methyl27-20160128_assays 76.8 Mb
## 154 EH2157 KIRC_Methylation_methyl27-20160128_se 0.4 Mb
## 155 EH2158 KIRC_Methylation_methyl450-20160128_assays 1418.8 Mb
## 156 EH2159 KIRC_Methylation_methyl450-20160128_se 6.1 Mb
## 157 EH720 KIRC_miRNASeqGene-20160128 0.2 Mb
## 158 EH721 KIRC_mRNAArray-20160128 3.5 Mb
## 159 EH722 KIRC_Mutation-20160128 0.4 Mb
## 160 EH723 KIRC_RNASeq2GeneNorm-20160128 32.2 Mb
## 161 EH724 KIRC_RNASeqGene-20160128 18.2 Mb
## 162 EH725 KIRC_RPPAArray-20160128 0.8 Mb
## 163 EH727 KIRP_CNASNP-20160128 2.6 Mb
## 164 EH728 KIRP_CNVSNP-20160128 0.5 Mb
## 165 EH730 KIRP_GISTIC_AllByGene-20160128 0.3 Mb
## 166 EH2160 KIRP_GISTIC_Peaks-20160128 0 Mb
## 167 EH731 KIRP_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 168 EH2161 KIRP_Methylation_methyl27-20160128_assays 3.9 Mb
## 169 EH2162 KIRP_Methylation_methyl27-20160128_se 0.4 Mb
## 170 EH2163 KIRP_Methylation_methyl450-20160128_assays 948 Mb
## 171 EH2164 KIRP_Methylation_methyl450-20160128_se 6.1 Mb
## 172 EH735 KIRP_miRNASeqGene-20160128 0.3 Mb
## 173 EH736 KIRP_mRNAArray-20160128 0.9 Mb
## 174 EH737 KIRP_Mutation-20160128 0.5 Mb
## 175 EH738 KIRP_RNASeq2GeneNorm-20160128 16.7 Mb
## 176 EH739 KIRP_RNASeqGene-20160128 0.6 Mb
## 177 EH740 KIRP_RPPAArray-20160128 0.3 Mb
## 178 EH742 LAML_CNASNP-20160128 7.6 Mb
## 179 EH743 LAML_CNVSNP-20160128 0.3 Mb
## 180 EH2538 LAML_GISTIC_AllByGene-20160128 0.3 Mb
## 181 EH2539 LAML_GISTIC_Peaks-20160128 0 Mb
## 182 EH2540 LAML_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 183 EH2166 LAML_Methylation_methyl27-20160128_assays 35.6 Mb
## 184 EH2167 LAML_Methylation_methyl27-20160128_se 0.4 Mb
## 185 EH2168 LAML_Methylation_methyl450-20160128_assays 572.8 Mb
## 186 EH2169 LAML_Methylation_methyl450-20160128_se 6.1 Mb
## 187 EH748 LAML_Mutation-20160128 0.2 Mb
## 188 EH749 LAML_RNASeq2GeneNorm-20160128 8.1 Mb
## 189 EH750 LAML_RNASeqGene-20160128 5.4 Mb
## 190 EH752 LGG_CNASeq-20160128 0.1 Mb
## 191 EH753 LGG_CNASNP-20160128 3.3 Mb
## 192 EH754 LGG_CNVSNP-20160128 0.8 Mb
## 193 EH756 LGG_GISTIC_AllByGene-20160128 0.5 Mb
## 194 EH2170 LGG_GISTIC_Peaks-20160128 0 Mb
## 195 EH757 LGG_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 196 EH2171 LGG_Methylation-20160128_assays 1564.1 Mb
## 197 EH2172 LGG_Methylation-20160128_se 6.1 Mb
## 198 EH760 LGG_miRNASeqGene-20160128 0.4 Mb
## 199 EH761 LGG_mRNAArray-20160128 1.7 Mb
## 200 EH762 LGG_Mutation-20160128 0.2 Mb
## 201 EH763 LGG_RNASeq2GeneNorm-20160128 28.3 Mb
## 202 EH764 LGG_RPPAArray-20160128 0.6 Mb
## 203 EH766 LIHC_CNASNP-20160128 3.1 Mb
## 204 EH767 LIHC_CNVSNP-20160128 1 Mb
## 205 EH769 LIHC_GISTIC_AllByGene-20160128 0.6 Mb
## 206 EH2173 LIHC_GISTIC_Peaks-20160128 0 Mb
## 207 EH770 LIHC_GISTIC_ThresholdedByGene-20160128 0.3 Mb
## 208 EH2174 LIHC_Methylation-20160128_assays 1267.8 Mb
## 209 EH2175 LIHC_Methylation-20160128_se 6.1 Mb
## 210 EH773 LIHC_miRNASeqGene-20160128 0.3 Mb
## 211 EH774 LIHC_Mutation-20160128 0.9 Mb
## 212 EH775 LIHC_RNASeq2GeneNorm-20160128 20.6 Mb
## 213 EH776 LIHC_RNASeqGene-20160128 0.9 Mb
## 214 EH777 LIHC_RPPAArray-20160128 0.3 Mb
## 215 EH779 LUAD_CNASeq-20160128 3.8 Mb
## 216 EH780 LUAD_CNASNP-20160128 4.2 Mb
## 217 EH781 LUAD_CNVSNP-20160128 1.2 Mb
## 218 EH783 LUAD_GISTIC_AllByGene-20160128 0.7 Mb
## 219 EH2176 LUAD_GISTIC_Peaks-20160128 0 Mb
## 220 EH784 LUAD_GISTIC_ThresholdedByGene-20160128 0.3 Mb
## 221 EH2177 LUAD_Methylation_methyl27-20160128_assays 16.4 Mb
## 222 EH2178 LUAD_Methylation_methyl27-20160128_se 0.4 Mb
## 223 EH2179 LUAD_Methylation_methyl450-20160128_assays 1452.7 Mb
## 224 EH2180 LUAD_Methylation_methyl450-20160128_se 6.1 Mb
## 225 EH788 LUAD_miRNASeqGene-20160128 0.4 Mb
## 226 EH789 LUAD_mRNAArray-20160128 1.6 Mb
## 227 EH790 LUAD_Mutation-20160128 6.7 Mb
## 228 EH791 LUAD_RNASeq2GeneNorm-20160128 29.8 Mb
## 229 EH792 LUAD_RNASeqGene-20160128 5.6 Mb
## 230 EH793 LUAD_RPPAArray-20160128 0.6 Mb
## 231 EH795 LUSC_CNACGH-20160128 0.7 Mb
## 232 EH796 LUSC_CNASNP-20160128 4.7 Mb
## 233 EH797 LUSC_CNVSNP-20160128 1.4 Mb
## 234 EH799 LUSC_GISTIC_AllByGene-20160128 0.7 Mb
## 235 EH2181 LUSC_GISTIC_Peaks-20160128 0 Mb
## 236 EH800 LUSC_GISTIC_ThresholdedByGene-20160128 0.3 Mb
## 237 EH2182 LUSC_Methylation_methyl27-20160128_assays 29.5 Mb
## 238 EH2183 LUSC_Methylation_methyl27-20160128_se 0.4 Mb
## 239 EH2184 LUSC_Methylation_methyl450-20160128_assays 1218.4 Mb
## 240 EH2185 LUSC_Methylation_methyl450-20160128_se 6.1 Mb
## 241 EH804 LUSC_miRNASeqGene-20160128 0.3 Mb
## 242 EH805 LUSC_mRNAArray_huex-20160128 14.8 Mb
## 243 EH806 LUSC_mRNAArray_TX_g4502a-20160128 7.2 Mb
## 244 EH807 LUSC_mRNAArray_TX_ht_hg_u133a-20160128 11.4 Mb
## 245 EH808 LUSC_Mutation-20160128 5.7 Mb
## 246 EH809 LUSC_RNASeq2GeneNorm-20160128 29.2 Mb
## 247 EH810 LUSC_RNASeqGene-20160128 8.4 Mb
## 248 EH811 LUSC_RPPAArray-20160128 0.5 Mb
## 249 EH813 MESO_CNASNP-20160128 0.8 Mb
## 250 EH814 MESO_CNVSNP-20160128 0.2 Mb
## 251 EH816 MESO_GISTIC_AllByGene-20160128 0.3 Mb
## 252 EH2186 MESO_GISTIC_Peaks-20160128 0 Mb
## 253 EH817 MESO_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 254 EH2187 MESO_Methylation-20160128_assays 257.2 Mb
## 255 EH2188 MESO_Methylation-20160128_se 6.1 Mb
## 256 EH820 MESO_miRNASeqGene-20160128 0.1 Mb
## 257 EH821 MESO_RNASeq2GeneNorm-20160128 4.6 Mb
## 258 EH822 MESO_RPPAArray-20160128 0.1 Mb
## 259 EH824 OV_CNACGH_CGH_hg_244a-20160128 1.1 Mb
## 260 EH825 OV_CNACGH_CGH_hg_415k_g4124a-20160128 2.2 Mb
## 261 EH826 OV_CNASNP-20160128 8.2 Mb
## 262 EH827 OV_CNVSNP-20160128 2.7 Mb
## 263 EH829 OV_GISTIC_AllByGene-20160128 1.2 Mb
## 264 EH2189 OV_GISTIC_Peaks-20160128 0 Mb
## 265 EH830 OV_GISTIC_ThresholdedByGene-20160128 0.4 Mb
## 266 EH2190 OV_Methylation_methyl27-20160128_assays 108.7 Mb
## 267 EH2191 OV_Methylation_methyl27-20160128_se 0.4 Mb
## 268 EH2192 OV_Methylation_methyl450-20160128_assays 29.6 Mb
## 269 EH2193 OV_Methylation_methyl450-20160128_se 6.1 Mb
## 270 EH834 OV_miRNAArray-20160128 3.2 Mb
## 271 EH835 OV_miRNASeqGene-20160128 0.3 Mb
## 272 EH836 OV_mRNAArray_huex-20160128 75.2 Mb
## 273 EH2194 OV_mRNAArray_TX_g4502a_1-20160128 25.2 Mb
## 274 EH837 OV_mRNAArray_TX_g4502a-20160128 1.9 Mb
## 275 EH838 OV_mRNAArray_TX_ht_hg_u133a-20160128 44.6 Mb
## 276 EH839 OV_Mutation-20160128 0.5 Mb
## 277 EH840 OV_RNASeq2GeneNorm-20160128 16.7 Mb
## 278 EH841 OV_RNASeqGene-20160128 10.4 Mb
## 279 EH842 OV_RPPAArray-20160128 0.7 Mb
## 280 EH844 PAAD_CNASNP-20160128 1.7 Mb
## 281 EH845 PAAD_CNVSNP-20160128 0.4 Mb
## 282 EH847 PAAD_GISTIC_AllByGene-20160128 0.4 Mb
## 283 EH2195 PAAD_GISTIC_Peaks-20160128 0 Mb
## 284 EH848 PAAD_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 285 EH2196 PAAD_Methylation-20160128_assays 575.8 Mb
## 286 EH2197 PAAD_Methylation-20160128_se 6.1 Mb
## 287 EH851 PAAD_miRNASeqGene-20160128 0.2 Mb
## 288 EH852 PAAD_Mutation-20160128 6.8 Mb
## 289 EH853 PAAD_RNASeq2GeneNorm-20160128 9.7 Mb
## 290 EH854 PAAD_RPPAArray-20160128 0.2 Mb
## 291 EH856 PCPG_CNASNP-20160128 2.6 Mb
## 292 EH857 PCPG_CNVSNP-20160128 0.3 Mb
## 293 EH859 PCPG_GISTIC_AllByGene-20160128 0.3 Mb
## 294 EH2198 PCPG_GISTIC_Peaks-20160128 0 Mb
## 295 EH860 PCPG_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 296 EH2199 PCPG_Methylation-20160128_assays 552.9 Mb
## 297 EH2200 PCPG_Methylation-20160128_se 6.1 Mb
## 298 EH863 PCPG_miRNASeqGene-20160128 0.2 Mb
## 299 EH864 PCPG_Mutation-20160128 0.6 Mb
## 300 EH865 PCPG_RNASeq2GeneNorm-20160128 9.6 Mb
## 301 EH866 PCPG_RPPAArray-20160128 0.1 Mb
## 302 EH868 PRAD_CNASeq-20160128 0.2 Mb
## 303 EH869 PRAD_CNASNP-20160128 4.9 Mb
## 304 EH870 PRAD_CNVSNP-20160128 1.2 Mb
## 305 EH872 PRAD_GISTIC_AllByGene-20160128 0.6 Mb
## 306 EH2201 PRAD_GISTIC_Peaks-20160128 0 Mb
## 307 EH873 PRAD_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 308 EH2202 PRAD_Methylation-20160128_assays 1622.5 Mb
## 309 EH2203 PRAD_Methylation-20160128_se 6.1 Mb
## 310 EH876 PRAD_miRNASeqGene-20160128 0.4 Mb
## 311 EH877 PRAD_Mutation-20160128 1.4 Mb
## 312 EH878 PRAD_RNASeq2GeneNorm-20160128 28.9 Mb
## 313 EH879 PRAD_RPPAArray-20160128 0.5 Mb
## 314 EH881 READ_CNASeq-20160128 0.4 Mb
## 315 EH882 READ_CNASNP-20160128 1.4 Mb
## 316 EH883 READ_CNVSNP-20160128 0.4 Mb
## 317 EH885 READ_GISTIC_AllByGene-20160128 0.3 Mb
## 318 EH2204 READ_GISTIC_Peaks-20160128 0 Mb
## 319 EH886 READ_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 320 EH2205 READ_Methylation_methyl27-20160128_assays 13.5 Mb
## 321 EH2206 READ_Methylation_methyl27-20160128_se 0.4 Mb
## 322 EH2207 READ_Methylation_methyl450-20160128_assays 313.3 Mb
## 323 EH2208 READ_Methylation_methyl450-20160128_se 6.1 Mb
## 324 EH890 READ_miRNASeqGene-20160128 0.1 Mb
## 325 EH891 READ_mRNAArray-20160128 3.5 Mb
## 326 EH892 READ_Mutation-20160128 0.4 Mb
## 327 EH893 READ_RNASeq2GeneNorm-20160128 3.5 Mb
## 328 EH894 READ_RNASeqGene-20160128 2.1 Mb
## 329 EH895 READ_RPPAArray-20160128 0.2 Mb
## 330 EH897 SARC_CNASNP-20160128 3.1 Mb
## 331 EH898 SARC_CNVSNP-20160128 1.1 Mb
## 332 EH900 SARC_GISTIC_AllByGene-20160128 0.6 Mb
## 333 EH2209 SARC_GISTIC_Peaks-20160128 0 Mb
## 334 EH901 SARC_GISTIC_ThresholdedByGene-20160128 0.3 Mb
## 335 EH2210 SARC_Methylation-20160128_assays 794.3 Mb
## 336 EH2211 SARC_Methylation-20160128_se 6.1 Mb
## 337 EH904 SARC_miRNASeqGene-20160128 0.2 Mb
## 338 EH905 SARC_Mutation-20160128 1.1 Mb
## 339 EH906 SARC_RNASeq2GeneNorm-20160128 13.6 Mb
## 340 EH907 SARC_RPPAArray-20160128 0.3 Mb
## 341 EH1029 SKCM_CNASeq-20160128 0.3 Mb
## 342 EH1030 SKCM_CNASNP-20160128 3.8 Mb
## 343 EH1031 SKCM_CNVSNP-20160128 1.1 Mb
## 344 EH2541 SKCM_GISTIC_AllByGene-20160128 0.5 Mb
## 345 EH2542 SKCM_GISTIC_Peaks-20160128 0 Mb
## 346 EH2543 SKCM_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 347 EH2213 SKCM_Methylation-20160128_assays 1403.5 Mb
## 348 EH2214 SKCM_Methylation-20160128_se 6.1 Mb
## 349 EH1033 SKCM_miRNASeqGene-20160128 0.4 Mb
## 350 EH1034 SKCM_Mutation-20160128 26.7 Mb
## 351 EH1035 SKCM_RNASeq2GeneNorm-20160128 24.5 Mb
## 352 EH1036 SKCM_RPPAArray-20160128 0.6 Mb
## 353 EH920 STAD_CNASeq-20160128 0.3 Mb
## 354 EH921 STAD_CNASNP-20160128 3.8 Mb
## 355 EH922 STAD_CNVSNP-20160128 1.2 Mb
## 356 EH924 STAD_GISTIC_AllByGene-20160128 0.7 Mb
## 357 EH2215 STAD_GISTIC_Peaks-20160128 0 Mb
## 358 EH925 STAD_GISTIC_ThresholdedByGene-20160128 0.3 Mb
## 359 EH2216 STAD_Methylation_methyl27-20160128_assays 13.5 Mb
## 360 EH2217 STAD_Methylation_methyl27-20160128_se 0.4 Mb
## 361 EH2218 STAD_Methylation_methyl450-20160128_assays 1172.9 Mb
## 362 EH2219 STAD_Methylation_methyl450-20160128_se 6.1 Mb
## 363 EH929 STAD_miRNASeqGene-20160128 0.3 Mb
## 364 EH930 STAD_Mutation-20160128 13.2 Mb
## 365 EH931 STAD_RNASeq2GeneNorm-20160128 23.9 Mb
## 366 EH932 STAD_RNASeqGene-20160128 1.6 Mb
## 367 EH933 STAD_RPPAArray-20160128 0.5 Mb
## 368 EH935 TGCT_CNASNP-20160128 1.2 Mb
## 369 EH936 TGCT_CNVSNP-20160128 0.3 Mb
## 370 EH938 TGCT_GISTIC_AllByGene-20160128 0.3 Mb
## 371 EH2220 TGCT_GISTIC_Peaks-20160128 0 Mb
## 372 EH939 TGCT_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 373 EH2221 TGCT_Methylation-20160128_assays 411.3 Mb
## 374 EH2222 TGCT_Methylation-20160128_se 6.1 Mb
## 375 EH942 TGCT_miRNASeqGene-20160128 0.2 Mb
## 376 EH943 TGCT_Mutation-20160128 0.5 Mb
## 377 EH944 TGCT_RNASeq2GeneNorm-20160128 7.4 Mb
## 378 EH945 TGCT_RPPAArray-20160128 0.2 Mb
## 379 EH947 THCA_CNASeq-20160128 0 Mb
## 380 EH948 THCA_CNASNP-20160128 3.1 Mb
## 381 EH949 THCA_CNVSNP-20160128 0.5 Mb
## 382 EH951 THCA_GISTIC_AllByGene-20160128 0.3 Mb
## 383 EH2223 THCA_GISTIC_Peaks-20160128 0 Mb
## 384 EH952 THCA_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 385 EH2224 THCA_Methylation-20160128_assays 1674.6 Mb
## 386 EH2225 THCA_Methylation-20160128_se 6.1 Mb
## 387 EH955 THCA_miRNASeqGene-20160128 0.5 Mb
## 388 EH956 THCA_Mutation-20160128 0.9 Mb
## 389 EH957 THCA_RNASeq2GeneNorm-20160128 30.1 Mb
## 390 EH958 THCA_RNASeqGene-20160128 0.2 Mb
## 391 EH959 THCA_RPPAArray-20160128 0.3 Mb
## 392 EH961 THYM_CNASNP-20160128 0.9 Mb
## 393 EH962 THYM_CNVSNP-20160128 0.2 Mb
## 394 EH964 THYM_GISTIC_AllByGene-20160128 0.2 Mb
## 395 EH2226 THYM_GISTIC_Peaks-20160128 0 Mb
## 396 EH965 THYM_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 397 EH2227 THYM_Methylation-20160128_assays 372.2 Mb
## 398 EH2228 THYM_Methylation-20160128_se 6.1 Mb
## 399 EH968 THYM_miRNASeqGene-20160128 0.1 Mb
## 400 EH969 THYM_Mutation-20160128 0.2 Mb
## 401 EH970 THYM_RNASeq2GeneNorm-20160128 6.4 Mb
## 402 EH971 THYM_RPPAArray-20160128 0.2 Mb
## 403 EH973 UCEC_CNASeq-20160128 0.3 Mb
## 404 EH974 UCEC_CNASNP-20160128 5.4 Mb
## 405 EH975 UCEC_CNVSNP-20160128 1.3 Mb
## 406 EH977 UCEC_GISTIC_AllByGene-20160128 0.7 Mb
## 407 EH2229 UCEC_GISTIC_Peaks-20160128 0 Mb
## 408 EH978 UCEC_GISTIC_ThresholdedByGene-20160128 0.3 Mb
## 409 EH2230 UCEC_Methylation_methyl27-20160128_assays 21.7 Mb
## 410 EH2231 UCEC_Methylation_methyl27-20160128_se 0.4 Mb
## 411 EH2232 UCEC_Methylation_methyl450-20160128_assays 1377 Mb
## 412 EH2233 UCEC_Methylation_methyl450-20160128_se 6.1 Mb
## 413 EH982 UCEC_miRNASeqGene-20160128 0.4 Mb
## 414 EH983 UCEC_mRNAArray-20160128 2.6 Mb
## 415 EH984 UCEC_Mutation-20160128 4.6 Mb
## 416 EH985 UCEC_RNASeq2GeneNorm-20160128 18.4 Mb
## 417 EH986 UCEC_RNASeqGene-20160128 7.7 Mb
## 418 EH987 UCEC_RPPAArray-20160128 0.7 Mb
## 419 EH989 UCS_CNASNP-20160128 0.5 Mb
## 420 EH990 UCS_CNVSNP-20160128 0.2 Mb
## 421 EH992 UCS_GISTIC_AllByGene-20160128 0.3 Mb
## 422 EH2234 UCS_GISTIC_Peaks-20160128 0 Mb
## 423 EH993 UCS_GISTIC_ThresholdedByGene-20160128 0.3 Mb
## 424 EH2235 UCS_Methylation-20160128_assays 168.3 Mb
## 425 EH2236 UCS_Methylation-20160128_se 6.1 Mb
## 426 EH996 UCS_miRNASeqGene-20160128 0.1 Mb
## 427 EH997 UCS_Mutation-20160128 1.2 Mb
## 428 EH998 UCS_RNASeq2GeneNorm-20160128 3.1 Mb
## 429 EH999 UCS_RPPAArray-20160128 0.1 Mb
## 430 EH1001 UVM_CNASeq-20160128 0.1 Mb
## 431 EH1002 UVM_CNASNP-20160128 0.6 Mb
## 432 EH1003 UVM_CNVSNP-20160128 0.1 Mb
## 433 EH1005 UVM_GISTIC_AllByGene-20160128 0.3 Mb
## 434 EH2237 UVM_GISTIC_Peaks-20160128 0 Mb
## 435 EH1006 UVM_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 436 EH2238 UVM_Methylation-20160128_assays 236.4 Mb
## 437 EH2239 UVM_Methylation-20160128_se 6.1 Mb
## 438 EH1009 UVM_miRNASeqGene-20160128 0.1 Mb
## 439 EH1010 UVM_Mutation-20160128 0.9 Mb
## 440 EH1011 UVM_RNASeq2GeneNorm-20160128 4 Mb
## 441 EH1012 UVM_RPPAArray-20160128 0 Mb
## rdataclass rdatadateadded rdatadateremoved
## 1 RaggedExperiment 2017-10-10 <NA>
## 2 RaggedExperiment 2017-10-10 <NA>
## 3 SummarizedExperiment 2017-10-10 <NA>
## 4 RangedSummarizedExperiment 2019-01-09 <NA>
## 5 SummarizedExperiment 2017-10-10 <NA>
## 6 SummarizedExperiment 2019-01-09 <NA>
## 7 RaggedExperiment 2019-01-09 <NA>
## 8 SummarizedExperiment 2017-10-10 <NA>
## 9 RaggedExperiment 2017-10-10 <NA>
## 10 SummarizedExperiment 2017-10-10 <NA>
## 11 SummarizedExperiment 2017-10-10 <NA>
## 12 RaggedExperiment 2017-10-10 <NA>
## 13 RaggedExperiment 2017-10-10 <NA>
## 14 RaggedExperiment 2017-10-10 <NA>
## 15 SummarizedExperiment 2017-10-10 <NA>
## 16 RangedSummarizedExperiment 2019-01-09 <NA>
## 17 SummarizedExperiment 2017-10-10 <NA>
## 18 SummarizedExperiment 2019-01-09 <NA>
## 19 RaggedExperiment 2019-01-09 <NA>
## 20 SummarizedExperiment 2017-10-10 <NA>
## 21 RaggedExperiment 2017-10-10 <NA>
## 22 SummarizedExperiment 2017-10-10 <NA>
## 23 SummarizedExperiment 2017-10-10 <NA>
## 24 SummarizedExperiment 2017-10-10 <NA>
## 25 RaggedExperiment 2017-10-10 <NA>
## 26 RaggedExperiment 2017-10-10 <NA>
## 27 RaggedExperiment 2017-10-10 <NA>
## 28 SummarizedExperiment 2017-10-10 <NA>
## 29 RangedSummarizedExperiment 2019-01-09 <NA>
## 30 SummarizedExperiment 2017-10-10 <NA>
## 31 SummarizedExperiment 2019-01-09 <NA>
## 32 SummarizedExperiment 2019-01-09 <NA>
## 33 RaggedExperiment 2019-01-09 <NA>
## 34 SummarizedExperiment 2019-01-09 <NA>
## 35 SummarizedExperiment 2017-10-10 <NA>
## 36 SummarizedExperiment 2017-10-10 <NA>
## 37 RaggedExperiment 2017-10-10 <NA>
## 38 SummarizedExperiment 2017-10-10 <NA>
## 39 SummarizedExperiment 2017-10-10 <NA>
## 40 SummarizedExperiment 2017-10-10 <NA>
## 41 RaggedExperiment 2017-10-10 <NA>
## 42 RaggedExperiment 2017-10-10 <NA>
## 43 RaggedExperiment 2017-10-10 <NA>
## 44 SummarizedExperiment 2017-10-10 <NA>
## 45 RangedSummarizedExperiment 2019-01-09 <NA>
## 46 SummarizedExperiment 2017-10-10 <NA>
## 47 SummarizedExperiment 2019-01-09 <NA>
## 48 RaggedExperiment 2019-01-09 <NA>
## 49 SummarizedExperiment 2017-10-10 <NA>
## 50 RaggedExperiment 2017-10-10 <NA>
## 51 SummarizedExperiment 2017-10-10 <NA>
## 52 SummarizedExperiment 2017-10-10 <NA>
## 53 RaggedExperiment 2017-10-10 <NA>
## 54 RaggedExperiment 2017-10-10 <NA>
## 55 SummarizedExperiment 2017-10-10 <NA>
## 56 RangedSummarizedExperiment 2019-01-09 <NA>
## 57 SummarizedExperiment 2017-10-10 <NA>
## 58 SummarizedExperiment 2019-01-09 <NA>
## 59 RaggedExperiment 2019-01-09 <NA>
## 60 SummarizedExperiment 2017-10-10 <NA>
## 61 RaggedExperiment 2017-10-10 <NA>
## 62 SummarizedExperiment 2017-10-10 <NA>
## 63 SummarizedExperiment 2017-10-10 <NA>
## 64 RaggedExperiment 2017-10-10 <NA>
## 65 RaggedExperiment 2017-10-10 <NA>
## 66 RaggedExperiment 2017-10-10 <NA>
## 67 SummarizedExperiment 2017-10-10 <NA>
## 68 RangedSummarizedExperiment 2019-01-09 <NA>
## 69 SummarizedExperiment 2017-10-10 <NA>
## 70 SummarizedExperiment 2019-01-09 <NA>
## 71 SummarizedExperiment 2019-01-09 <NA>
## 72 RaggedExperiment 2019-01-09 <NA>
## 73 SummarizedExperiment 2019-01-09 <NA>
## 74 SummarizedExperiment 2017-10-10 <NA>
## 75 SummarizedExperiment 2017-10-10 <NA>
## 76 RaggedExperiment 2017-10-10 <NA>
## 77 SummarizedExperiment 2017-10-10 <NA>
## 78 SummarizedExperiment 2017-10-10 <NA>
## 79 SummarizedExperiment 2017-10-10 <NA>
## 80 RaggedExperiment 2017-10-10 <NA>
## 81 RaggedExperiment 2017-10-10 <NA>
## 82 SummarizedExperiment 2017-10-10 <NA>
## 83 RangedSummarizedExperiment 2019-01-09 <NA>
## 84 SummarizedExperiment 2017-10-10 <NA>
## 85 SummarizedExperiment 2019-01-09 <NA>
## 86 RaggedExperiment 2019-01-09 <NA>
## 87 SummarizedExperiment 2017-10-10 <NA>
## 88 RaggedExperiment 2017-10-10 <NA>
## 89 SummarizedExperiment 2017-10-10 <NA>
## 90 SummarizedExperiment 2017-10-10 <NA>
## 91 RaggedExperiment 2017-10-10 <NA>
## 92 RaggedExperiment 2017-10-10 <NA>
## 93 RaggedExperiment 2017-10-10 <NA>
## 94 SummarizedExperiment 2017-10-10 <NA>
## 95 RangedSummarizedExperiment 2019-01-09 <NA>
## 96 SummarizedExperiment 2017-10-10 <NA>
## 97 SummarizedExperiment 2019-01-09 <NA>
## 98 RaggedExperiment 2019-01-09 <NA>
## 99 SummarizedExperiment 2017-10-10 <NA>
## 100 RaggedExperiment 2017-10-10 <NA>
## 101 SummarizedExperiment 2017-10-10 <NA>
## 102 SummarizedExperiment 2017-10-10 <NA>
## 103 SummarizedExperiment 2017-10-10 <NA>
## 104 RaggedExperiment 2017-10-10 <NA>
## 105 RaggedExperiment 2017-10-10 <NA>
## 106 RaggedExperiment 2017-10-10 <NA>
## 107 RaggedExperiment 2017-10-10 <NA>
## 108 SummarizedExperiment 2017-10-10 <NA>
## 109 RangedSummarizedExperiment 2019-01-09 <NA>
## 110 SummarizedExperiment 2017-10-10 <NA>
## 111 SummarizedExperiment 2019-01-09 <NA>
## 112 SummarizedExperiment 2019-01-09 <NA>
## 113 SummarizedExperiment 2019-01-09 <NA>
## 114 SummarizedExperiment 2019-01-09 <NA>
## 115 SummarizedExperiment 2017-10-10 <NA>
## 116 SummarizedExperiment 2017-10-10 <NA>
## 117 SummarizedExperiment 2017-10-10 <NA>
## 118 SummarizedExperiment 2019-01-09 <NA>
## 119 SummarizedExperiment 2017-10-10 <NA>
## 120 SummarizedExperiment 2017-10-10 <NA>
## 121 RaggedExperiment 2017-10-10 <NA>
## 122 SummarizedExperiment 2017-10-10 <NA>
## 123 SummarizedExperiment 2017-10-10 <NA>
## 124 RaggedExperiment 2017-10-10 <NA>
## 125 RaggedExperiment 2017-10-10 <NA>
## 126 RaggedExperiment 2017-10-10 <NA>
## 127 SummarizedExperiment 2017-10-10 <NA>
## 128 RangedSummarizedExperiment 2019-01-09 <NA>
## 129 SummarizedExperiment 2017-10-10 <NA>
## 130 SummarizedExperiment 2019-01-09 <NA>
## 131 RaggedExperiment 2019-01-09 <NA>
## 132 SummarizedExperiment 2017-10-10 <NA>
## 133 RaggedExperiment 2017-10-10 <NA>
## 134 SummarizedExperiment 2017-10-10 <NA>
## 135 SummarizedExperiment 2017-10-10 <NA>
## 136 SummarizedExperiment 2017-10-10 <NA>
## 137 RaggedExperiment 2017-10-10 <NA>
## 138 RaggedExperiment 2017-10-10 <NA>
## 139 SummarizedExperiment 2017-10-10 <NA>
## 140 RangedSummarizedExperiment 2019-01-09 <NA>
## 141 SummarizedExperiment 2017-10-10 <NA>
## 142 SummarizedExperiment 2019-01-09 <NA>
## 143 RaggedExperiment 2019-01-09 <NA>
## 144 SummarizedExperiment 2017-10-10 <NA>
## 145 RaggedExperiment 2017-10-10 <NA>
## 146 SummarizedExperiment 2017-10-10 <NA>
## 147 SummarizedExperiment 2017-10-10 <NA>
## 148 RaggedExperiment 2017-10-10 <NA>
## 149 RaggedExperiment 2017-10-10 <NA>
## 150 SummarizedExperiment 2017-10-10 <NA>
## 151 RangedSummarizedExperiment 2019-01-09 <NA>
## 152 SummarizedExperiment 2017-10-10 <NA>
## 153 SummarizedExperiment 2019-01-09 <NA>
## 154 SummarizedExperiment 2019-01-09 <NA>
## 155 RaggedExperiment 2019-01-09 <NA>
## 156 SummarizedExperiment 2019-01-09 <NA>
## 157 SummarizedExperiment 2017-10-10 <NA>
## 158 SummarizedExperiment 2017-10-10 <NA>
## 159 RaggedExperiment 2017-10-10 <NA>
## 160 SummarizedExperiment 2017-10-10 <NA>
## 161 SummarizedExperiment 2017-10-10 <NA>
## 162 SummarizedExperiment 2017-10-10 <NA>
## 163 RaggedExperiment 2017-10-10 <NA>
## 164 RaggedExperiment 2017-10-10 <NA>
## 165 SummarizedExperiment 2017-10-10 <NA>
## 166 RangedSummarizedExperiment 2019-01-09 <NA>
## 167 SummarizedExperiment 2017-10-10 <NA>
## 168 SummarizedExperiment 2019-01-09 <NA>
## 169 SummarizedExperiment 2019-01-09 <NA>
## 170 RaggedExperiment 2019-01-09 <NA>
## 171 SummarizedExperiment 2019-01-09 <NA>
## 172 SummarizedExperiment 2017-10-10 <NA>
## 173 SummarizedExperiment 2017-10-10 <NA>
## 174 RaggedExperiment 2017-10-10 <NA>
## 175 SummarizedExperiment 2017-10-10 <NA>
## 176 SummarizedExperiment 2017-10-10 <NA>
## 177 SummarizedExperiment 2017-10-10 <NA>
## 178 RaggedExperiment 2017-10-10 <NA>
## 179 RaggedExperiment 2017-10-10 <NA>
## 180 SummarizedExperiment 2019-04-29 <NA>
## 181 RangedSummarizedExperiment 2019-04-29 <NA>
## 182 SummarizedExperiment 2019-04-29 <NA>
## 183 RaggedExperiment 2019-01-09 <NA>
## 184 SummarizedExperiment 2019-01-09 <NA>
## 185 SummarizedExperiment 2019-01-09 <NA>
## 186 DataFrame 2019-01-09 <NA>
## 187 RaggedExperiment 2017-10-10 <NA>
## 188 SummarizedExperiment 2017-10-10 <NA>
## 189 SummarizedExperiment 2017-10-10 <NA>
## 190 RaggedExperiment 2017-10-10 <NA>
## 191 RaggedExperiment 2017-10-10 <NA>
## 192 RaggedExperiment 2017-10-10 <NA>
## 193 SummarizedExperiment 2017-10-10 <NA>
## 194 RangedSummarizedExperiment 2019-01-09 <NA>
## 195 SummarizedExperiment 2017-10-10 <NA>
## 196 SummarizedExperiment 2019-01-09 <NA>
## 197 SummarizedExperiment 2019-01-09 <NA>
## 198 SummarizedExperiment 2017-10-10 <NA>
## 199 SummarizedExperiment 2017-10-10 <NA>
## 200 RaggedExperiment 2017-10-10 <NA>
## 201 SummarizedExperiment 2017-10-10 <NA>
## 202 SummarizedExperiment 2017-10-10 <NA>
## 203 RaggedExperiment 2017-10-10 <NA>
## 204 RaggedExperiment 2017-10-10 <NA>
## 205 SummarizedExperiment 2017-10-10 <NA>
## 206 RangedSummarizedExperiment 2019-01-09 <NA>
## 207 SummarizedExperiment 2017-10-10 <NA>
## 208 SummarizedExperiment 2019-01-09 <NA>
## 209 RaggedExperiment 2019-01-09 <NA>
## 210 SummarizedExperiment 2017-10-10 <NA>
## 211 RaggedExperiment 2017-10-10 <NA>
## 212 SummarizedExperiment 2017-10-10 <NA>
## 213 SummarizedExperiment 2017-10-10 <NA>
## 214 SummarizedExperiment 2017-10-10 <NA>
## 215 RaggedExperiment 2017-10-10 <NA>
## 216 RaggedExperiment 2017-10-10 <NA>
## 217 RaggedExperiment 2017-10-10 <NA>
## 218 SummarizedExperiment 2017-10-10 <NA>
## 219 RangedSummarizedExperiment 2019-01-09 <NA>
## 220 SummarizedExperiment 2017-10-10 <NA>
## 221 SummarizedExperiment 2019-01-09 <NA>
## 222 SummarizedExperiment 2019-01-09 <NA>
## 223 RaggedExperiment 2019-01-09 <NA>
## 224 SummarizedExperiment 2019-01-09 <NA>
## 225 SummarizedExperiment 2017-10-10 <NA>
## 226 SummarizedExperiment 2017-10-10 <NA>
## 227 RaggedExperiment 2017-10-10 <NA>
## 228 SummarizedExperiment 2017-10-10 <NA>
## 229 SummarizedExperiment 2017-10-10 <NA>
## 230 SummarizedExperiment 2017-10-10 <NA>
## 231 RaggedExperiment 2017-10-10 <NA>
## 232 RaggedExperiment 2017-10-10 <NA>
## 233 RaggedExperiment 2017-10-10 <NA>
## 234 SummarizedExperiment 2017-10-10 <NA>
## 235 RangedSummarizedExperiment 2019-01-09 <NA>
## 236 SummarizedExperiment 2017-10-10 <NA>
## 237 SummarizedExperiment 2019-01-09 <NA>
## 238 SummarizedExperiment 2019-01-09 <NA>
## 239 SummarizedExperiment 2019-01-09 <NA>
## 240 SummarizedExperiment 2019-01-09 <NA>
## 241 SummarizedExperiment 2017-10-10 <NA>
## 242 SummarizedExperiment 2017-10-10 <NA>
## 243 SummarizedExperiment 2017-10-10 <NA>
## 244 SummarizedExperiment 2017-10-10 <NA>
## 245 RaggedExperiment 2017-10-10 <NA>
## 246 SummarizedExperiment 2017-10-10 <NA>
## 247 SummarizedExperiment 2017-10-10 <NA>
## 248 SummarizedExperiment 2017-10-10 <NA>
## 249 RaggedExperiment 2017-10-10 <NA>
## 250 RaggedExperiment 2017-10-10 <NA>
## 251 SummarizedExperiment 2017-10-10 <NA>
## 252 RangedSummarizedExperiment 2019-01-09 <NA>
## 253 SummarizedExperiment 2017-10-10 <NA>
## 254 SummarizedExperiment 2019-01-09 <NA>
## 255 SummarizedExperiment 2019-01-09 <NA>
## 256 SummarizedExperiment 2017-10-10 <NA>
## 257 SummarizedExperiment 2017-10-10 <NA>
## 258 SummarizedExperiment 2017-10-10 <NA>
## 259 RaggedExperiment 2017-10-10 <NA>
## 260 RaggedExperiment 2017-10-10 <NA>
## 261 RaggedExperiment 2017-10-10 <NA>
## 262 RaggedExperiment 2017-10-10 <NA>
## 263 SummarizedExperiment 2017-10-10 <NA>
## 264 RangedSummarizedExperiment 2019-01-09 <NA>
## 265 SummarizedExperiment 2017-10-10 <NA>
## 266 SummarizedExperiment 2019-01-09 <NA>
## 267 SummarizedExperiment 2019-01-09 <NA>
## 268 SummarizedExperiment 2019-01-09 <NA>
## 269 SummarizedExperiment 2019-01-09 <NA>
## 270 SummarizedExperiment 2017-10-10 <NA>
## 271 SummarizedExperiment 2017-10-10 <NA>
## 272 SummarizedExperiment 2017-10-10 <NA>
## 273 SummarizedExperiment 2019-01-09 <NA>
## 274 SummarizedExperiment 2017-10-10 <NA>
## 275 SummarizedExperiment 2017-10-10 <NA>
## 276 RaggedExperiment 2017-10-10 <NA>
## 277 SummarizedExperiment 2017-10-10 <NA>
## 278 SummarizedExperiment 2017-10-10 <NA>
## 279 SummarizedExperiment 2017-10-10 <NA>
## 280 RaggedExperiment 2017-10-10 <NA>
## 281 RaggedExperiment 2017-10-10 <NA>
## 282 SummarizedExperiment 2017-10-10 <NA>
## 283 RangedSummarizedExperiment 2019-01-09 <NA>
## 284 SummarizedExperiment 2017-10-10 <NA>
## 285 SummarizedExperiment 2019-01-09 <NA>
## 286 RaggedExperiment 2019-01-09 <NA>
## 287 SummarizedExperiment 2017-10-10 <NA>
## 288 RaggedExperiment 2017-10-10 <NA>
## 289 SummarizedExperiment 2017-10-10 <NA>
## 290 SummarizedExperiment 2017-10-10 <NA>
## 291 RaggedExperiment 2017-10-10 <NA>
## 292 RaggedExperiment 2017-10-10 <NA>
## 293 SummarizedExperiment 2017-10-10 <NA>
## 294 RangedSummarizedExperiment 2019-01-09 <NA>
## 295 SummarizedExperiment 2017-10-10 <NA>
## 296 SummarizedExperiment 2019-01-09 <NA>
## 297 RaggedExperiment 2019-01-09 <NA>
## 298 SummarizedExperiment 2017-10-10 <NA>
## 299 RaggedExperiment 2017-10-10 <NA>
## 300 SummarizedExperiment 2017-10-10 <NA>
## 301 SummarizedExperiment 2017-10-10 <NA>
## 302 RaggedExperiment 2017-10-10 <NA>
## 303 RaggedExperiment 2017-10-10 <NA>
## 304 RaggedExperiment 2017-10-10 <NA>
## 305 SummarizedExperiment 2017-10-10 <NA>
## 306 RangedSummarizedExperiment 2019-01-09 <NA>
## 307 SummarizedExperiment 2017-10-10 <NA>
## 308 SummarizedExperiment 2019-01-09 <NA>
## 309 RaggedExperiment 2019-01-09 <NA>
## 310 SummarizedExperiment 2017-10-10 <NA>
## 311 RaggedExperiment 2017-10-10 <NA>
## 312 SummarizedExperiment 2017-10-10 <NA>
## 313 SummarizedExperiment 2017-10-10 <NA>
## 314 RaggedExperiment 2017-10-10 <NA>
## 315 RaggedExperiment 2017-10-10 <NA>
## 316 RaggedExperiment 2017-10-10 <NA>
## 317 SummarizedExperiment 2017-10-10 <NA>
## 318 RangedSummarizedExperiment 2019-01-09 <NA>
## 319 SummarizedExperiment 2017-10-10 <NA>
## 320 SummarizedExperiment 2019-01-09 <NA>
## 321 SummarizedExperiment 2019-01-09 <NA>
## 322 RaggedExperiment 2019-01-09 <NA>
## 323 SummarizedExperiment 2019-01-09 <NA>
## 324 SummarizedExperiment 2017-10-10 <NA>
## 325 SummarizedExperiment 2017-10-10 <NA>
## 326 RaggedExperiment 2017-10-10 <NA>
## 327 SummarizedExperiment 2017-10-10 <NA>
## 328 SummarizedExperiment 2017-10-10 <NA>
## 329 SummarizedExperiment 2017-10-10 <NA>
## 330 RaggedExperiment 2017-10-10 <NA>
## 331 RaggedExperiment 2017-10-10 <NA>
## 332 SummarizedExperiment 2017-10-10 <NA>
## 333 RangedSummarizedExperiment 2019-01-09 <NA>
## 334 SummarizedExperiment 2017-10-10 <NA>
## 335 SummarizedExperiment 2019-01-09 <NA>
## 336 RaggedExperiment 2019-01-09 <NA>
## 337 SummarizedExperiment 2017-10-10 <NA>
## 338 RaggedExperiment 2017-10-10 <NA>
## 339 SummarizedExperiment 2017-10-10 <NA>
## 340 SummarizedExperiment 2017-10-10 <NA>
## 341 RaggedExperiment 2017-10-16 <NA>
## 342 RaggedExperiment 2017-10-16 <NA>
## 343 RaggedExperiment 2017-10-16 <NA>
## 344 SummarizedExperiment 2019-04-29 <NA>
## 345 RangedSummarizedExperiment 2019-04-29 <NA>
## 346 SummarizedExperiment 2019-04-29 <NA>
## 347 SummarizedExperiment 2019-01-09 <NA>
## 348 RaggedExperiment 2019-01-09 <NA>
## 349 SummarizedExperiment 2017-10-16 <NA>
## 350 RaggedExperiment 2017-10-16 <NA>
## 351 SummarizedExperiment 2017-10-16 <NA>
## 352 SummarizedExperiment 2017-10-16 <NA>
## 353 RaggedExperiment 2017-10-10 <NA>
## 354 RaggedExperiment 2017-10-10 <NA>
## 355 RaggedExperiment 2017-10-10 <NA>
## 356 SummarizedExperiment 2017-10-10 <NA>
## 357 RangedSummarizedExperiment 2019-01-09 <NA>
## 358 SummarizedExperiment 2017-10-10 <NA>
## 359 SummarizedExperiment 2019-01-09 <NA>
## 360 RaggedExperiment 2019-01-09 <NA>
## 361 SummarizedExperiment 2019-01-09 <NA>
## 362 SummarizedExperiment 2019-01-09 <NA>
## 363 SummarizedExperiment 2017-10-10 <NA>
## 364 RaggedExperiment 2017-10-10 <NA>
## 365 SummarizedExperiment 2017-10-10 <NA>
## 366 SummarizedExperiment 2017-10-10 <NA>
## 367 SummarizedExperiment 2017-10-10 <NA>
## 368 RaggedExperiment 2017-10-10 <NA>
## 369 RaggedExperiment 2017-10-10 <NA>
## 370 SummarizedExperiment 2017-10-10 <NA>
## 371 RangedSummarizedExperiment 2019-01-09 <NA>
## 372 SummarizedExperiment 2017-10-10 <NA>
## 373 SummarizedExperiment 2019-01-09 <NA>
## 374 RaggedExperiment 2019-01-09 <NA>
## 375 SummarizedExperiment 2017-10-10 <NA>
## 376 RaggedExperiment 2017-10-10 <NA>
## 377 SummarizedExperiment 2017-10-10 <NA>
## 378 SummarizedExperiment 2017-10-10 <NA>
## 379 RaggedExperiment 2017-10-10 <NA>
## 380 RaggedExperiment 2017-10-10 <NA>
## 381 RaggedExperiment 2017-10-10 <NA>
## 382 SummarizedExperiment 2017-10-10 <NA>
## 383 RangedSummarizedExperiment 2019-01-09 <NA>
## 384 SummarizedExperiment 2017-10-10 <NA>
## 385 SummarizedExperiment 2019-01-09 <NA>
## 386 RaggedExperiment 2019-01-09 <NA>
## 387 SummarizedExperiment 2017-10-10 <NA>
## 388 RaggedExperiment 2017-10-10 <NA>
## 389 SummarizedExperiment 2017-10-10 <NA>
## 390 SummarizedExperiment 2017-10-10 <NA>
## 391 SummarizedExperiment 2017-10-10 <NA>
## 392 RaggedExperiment 2017-10-10 <NA>
## 393 RaggedExperiment 2017-10-10 <NA>
## 394 SummarizedExperiment 2017-10-10 <NA>
## 395 RangedSummarizedExperiment 2019-01-09 <NA>
## 396 SummarizedExperiment 2017-10-10 <NA>
## 397 SummarizedExperiment 2019-01-09 <NA>
## 398 RaggedExperiment 2019-01-09 <NA>
## 399 SummarizedExperiment 2017-10-10 <NA>
## 400 RaggedExperiment 2017-10-10 <NA>
## 401 SummarizedExperiment 2017-10-10 <NA>
## 402 SummarizedExperiment 2017-10-10 <NA>
## 403 RaggedExperiment 2017-10-10 <NA>
## 404 RaggedExperiment 2017-10-10 <NA>
## 405 RaggedExperiment 2017-10-10 <NA>
## 406 SummarizedExperiment 2017-10-10 <NA>
## 407 RangedSummarizedExperiment 2019-01-09 <NA>
## 408 SummarizedExperiment 2017-10-10 <NA>
## 409 SummarizedExperiment 2019-01-09 <NA>
## 410 SummarizedExperiment 2019-01-09 <NA>
## 411 RaggedExperiment 2019-01-09 <NA>
## 412 SummarizedExperiment 2019-01-09 <NA>
## 413 SummarizedExperiment 2017-10-10 <NA>
## 414 SummarizedExperiment 2017-10-10 <NA>
## 415 RaggedExperiment 2017-10-10 <NA>
## 416 SummarizedExperiment 2017-10-10 <NA>
## 417 SummarizedExperiment 2017-10-10 <NA>
## 418 SummarizedExperiment 2017-10-10 <NA>
## 419 RaggedExperiment 2017-10-10 <NA>
## 420 RaggedExperiment 2017-10-10 <NA>
## 421 SummarizedExperiment 2017-10-10 <NA>
## 422 RangedSummarizedExperiment 2019-01-09 <NA>
## 423 SummarizedExperiment 2017-10-10 <NA>
## 424 SummarizedExperiment 2019-01-09 <NA>
## 425 RaggedExperiment 2019-01-09 <NA>
## 426 SummarizedExperiment 2017-10-10 <NA>
## 427 RaggedExperiment 2017-10-10 <NA>
## 428 SummarizedExperiment 2017-10-10 <NA>
## 429 SummarizedExperiment 2017-10-10 <NA>
## 430 RaggedExperiment 2017-10-10 <NA>
## 431 RaggedExperiment 2017-10-10 <NA>
## 432 RaggedExperiment 2017-10-10 <NA>
## 433 SummarizedExperiment 2017-10-10 <NA>
## 434 RangedSummarizedExperiment 2019-01-09 <NA>
## 435 SummarizedExperiment 2017-10-10 <NA>
## 436 SummarizedExperiment 2019-01-09 <NA>
## 437 RaggedExperiment 2019-01-09 <NA>
## 438 SummarizedExperiment 2017-10-10 <NA>
## 439 RaggedExperiment 2017-10-10 <NA>
## 440 SummarizedExperiment 2017-10-10 <NA>
## 441 SummarizedExperiment 2017-10-10 <NA>Check potential files to be downloaded for adrenocortical carcinoma (ACC):
curatedTCGAData(diseaseCode = "ACC")## snapshotDate(): 2020-10-27## See '?curatedTCGAData' for 'diseaseCode' and 'assays' inputs## ah_id title file_size
## 1 EH558 ACC_CNASNP-20160128 0.8 Mb
## 2 EH559 ACC_CNVSNP-20160128 0.2 Mb
## 3 EH561 ACC_GISTIC_AllByGene-20160128 0.3 Mb
## 4 EH2115 ACC_GISTIC_Peaks-20160128 0 Mb
## 5 EH562 ACC_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 6 EH2116 ACC_Methylation-20160128_assays 236.4 Mb
## 7 EH2117 ACC_Methylation-20160128_se 6.1 Mb
## 8 EH565 ACC_miRNASeqGene-20160128 0.1 Mb
## 9 EH566 ACC_Mutation-20160128 0.7 Mb
## 10 EH567 ACC_RNASeq2GeneNorm-20160128 4 Mb
## 11 EH568 ACC_RPPAArray-20160128 0.1 Mb
## rdataclass rdatadateadded rdatadateremoved
## 1 RaggedExperiment 2017-10-10 <NA>
## 2 RaggedExperiment 2017-10-10 <NA>
## 3 SummarizedExperiment 2017-10-10 <NA>
## 4 RangedSummarizedExperiment 2019-01-09 <NA>
## 5 SummarizedExperiment 2017-10-10 <NA>
## 6 SummarizedExperiment 2019-01-09 <NA>
## 7 RaggedExperiment 2019-01-09 <NA>
## 8 SummarizedExperiment 2017-10-10 <NA>
## 9 RaggedExperiment 2017-10-10 <NA>
## 10 SummarizedExperiment 2017-10-10 <NA>
## 11 SummarizedExperiment 2017-10-10 <NA>Actually download the reverse phase protein array (RPPA) and RNA-seq data for ACC
ACCmae <- curatedTCGAData("ACC", c("RPPAArray", "RNASeq2GeneNorm"),
dry.run=FALSE)
ACCmae## A MultiAssayExperiment object of 2 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 2:
## [1] ACC_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 79 columns
## [2] ACC_RPPAArray-20160128: SummarizedExperiment with 192 rows and 46 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save all data to filesNote. Data will be downloaded the first time the above command is run; subsequent times it will be loaded from local cache.
This object contains 822 columns of clinical, pathological, specimen, and subtypes data in its colData, merged from all available data levels (1-4) of the Firehose pipeline:
## [1] 79 822## [1] "patientID" "years_to_birth" "vital_status"
## [4] "days_to_death" "days_to_last_followup" "tumor_tissue_site"See the MultiAssayExperiment vignette (Ramos et al. 2017) and the Workflow for Multi-omics Analysis with MultiAssayExperiment workshop (https://waldronlab.io/MultiAssayWorkshop/) for details on using this object.
Subtype information
Some cancer datasets contain associated subtype information within the clinical datasets provided. This subtype information is included in the metadata of colData of the MultiAssayExperiment object. To obtain these variable names, run the metadata function on the colData of the object such as:
## ACC_annotations ACC_subtype
## 1 Patient_ID SAMPLE
## 2 histological_subtypes Histology
## 3 mrna_subtypes C1A/C1B
## 4 mrna_subtypes mRNA_K4
## 5 cimp MethyLevel
## 6 microrna_subtypes miRNA clustercBioPortalData
cBioPortalData accesses most data from the Cancer Biology Portal cbioportal.org within R/Bioconductor using MultiAssayExperiment. It provides two main functions:
-
cBioDataPackfor pre-packaged full datasets -
cBioPortalDatafor API-query data
Interactive workshop: https://waldronlab.io/MultiAssayWorkshop/tutorials/cBioPortalData_Intro.html
recount: Reproducible RNA-seq Analysis Using recount2
The recount(Collado-Torres et al. 2017) package provides uniformly processed RangedSummarizedExperiment objects at the gene, exon, or exon-exon junctions level, the raw counts, the phenotype metadata used, the urls to sample coverage bigWig files and mean coverage bigWig file, for every study available. The RangedSummarizedExperiment objects can be used for differential expression analysis. These are also accessible through a web interface.6
recount provides a search function:
library(recount)
project_info <- abstract_search('GSE32465')It is not an ExperimentHub package, so downloading and serializing is slightly more involved in involves two steps: first, download the gene-level RangedSummarizedExperiment data:
download_study(project_info$project)## 2021-04-29 12:43:01 downloading file rse_gene.Rdata to SRP009615followed by loading the data
curated*Data packages for standardized cancer transcriptomes
There are focused databases of cancer microarray data for several cancer types, which can be useful for researchers of those cancer types or for methodological development:
- curatedOvarianData(Ganzfried et al. 2013): Clinically Annotated Data for the Ovarian Cancer Transcriptome (data available with additional options through the MetaGxOvarian package).
- curatedBladderData: Clinically Annotated Data for the Bladder Cancer Transcriptome
- curatedCRCData: Clinically Annotated Data for the Colorectal Cancer Transcriptome
These provide data from the Gene Expression Omnibus and other sources, but use a formally vocabulary for clinicopathological data and use a common pipeline for preprocessing of microarray data (for Affymetrix, other for other platforms the processed data are provided as processed by original study authors), merging probesets, and mapping to gene symbols. The pipeline is described by Ganzfried et al. (2013).
Microbiome data
Bioconductor provides curated resources of microbiome data. Most microbiome data are generated either by targeted amplicon sequencing (usually of variable regions of the 16S ribosomal RNA gene) or by metagenomic shotgun sequencing (MGX). These two approaches are analyzed by different sequence analysis tools, but downstream statistical and ecological analysis can involve any of the following types of data:
- taxonomic abundance at different levels of the taxonomic hierarchy
- phylogenetic distances and the phylogenetic tree of life
- metabolic potential of the microbiome
- abundance of microbial genes and gene families
A review of types and properties of microbiome data is provided by (Morgan and Huttenhower 2012).
curatedMetagenomicData: Curated and processed metagenomic data through ExperimentHub
curatedMetagenomicData(Pasolli et al. 2017) provides 6 types of processed data for >30 publicly available whole-metagenome shotgun sequencing datasets (obtained from the Sequence Read Archive):
- Species-level taxonomic profiles, expressed as relative abundance from kingdom to strain level
- Presence of unique, clade-specific markers
- Abundance of unique, clade-specific markers
- Abundance of gene families
- Metabolic pathway coverage
- Metabolic pathway abundance
Types 1-3 are generated by MetaPhlAn2; 4-6 are generated by HUMAnN2.
Currently, curatedMetagenomicData provides:
- 10199 samples from 57 datasets, primarily of the human gut but including body sites profiled in the Human Microbiome Project
- Processed data from whole-metagenome shotgun metagenomics, with manually-curated metadata, as integrated and documented Bioconductor ExpressionSet objects
- ~80 fields of specimen metadata from original papers, supplementary files, and websites, with manual curation to standardize annotations
- Processing of data through the MetaPhlAn2 pipeline for taxonomic abundance, and HUMAnN2 pipeline for metabolic analysis
- These represent ~100TB of raw sequencing data, but the processed data provided are much smaller.
These datasets are documented in the reference manual.
This is an ExperimentHub package, and its main workhorse function is curatedMetagenomicData():
The manually curated metadata for all available samples are provided in a single table combined_metadata:
library(curatedMetagenomicData)
?combined_metadata
View(data.frame(combined_metadata))The main function provides a list of ExpressionSet objects:
oral <- c("BritoIL_2016.metaphlan_bugs_list.oralcavity",
"Castro-NallarE_2015.metaphlan_bugs_list.oralcavity")
esl <- curatedMetagenomicData(oral, dryrun = FALSE)
esl## List of length 2
## names(2): BritoIL_2016.metaphlan_bugs_list.oralcavity ...These ExpressionSet objects can also be converted to phyloseq object for ecological analysis and differential abundance analysis using the DESeq2 package, using the ExpressionSet2phyloseq() function:
ExpressionSet2phyloseq( esl[[1]], phylogenetictree = TRUE)## Loading required namespace: phyloseq## Warning: `data_frame()` was deprecated in tibble 1.1.0.
## Please use `tibble()` instead.## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 535 taxa and 140 samples ]
## sample_data() Sample Data: [ 140 samples by 17 sample variables ]
## tax_table() Taxonomy Table: [ 535 taxa by 8 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 535 tips and 534 internal nodes ]See the documentation of phyloseq for more on ecological and differential abundance analysis of the microbiome.
HMP16SData: 16S rRNA Sequencing Data from the Human Microbiome Project
## snapshotDate(): 2020-10-27HMP16SData(Schiffer et al. 2018) is a Bioconductor ExperimentData package of the Human Microbiome Project (HMP) 16S rRNA sequencing data. Taxonomic count data files are provided as downloaded from the HMP Data Analysis and Coordination Center from its QIIME pipeline. Processed data is provided as SummarizedExperiment class objects via ExperimentHub. Like other ExperimentHub-based packages, a convenience function does downloading, automatic local caching, and serializing of a Bioconductor data class. This returns taxonomic counts from the V1-3 variable region of the 16S rRNA gene, along with the unrestricted participant data and phylogenetic tree.
V13()## class: SummarizedExperiment
## dim: 43140 2898
## metadata(2): experimentData phylogeneticTree
## assays(1): 16SrRNA
## rownames(43140): OTU_97.1 OTU_97.10 ... OTU_97.9997 OTU_97.9999
## rowData names(7): CONSENSUS_LINEAGE SUPERKINGDOM ... FAMILY GENUS
## colnames(2898): 700013549 700014386 ... 700114963 700114965
## colData names(7): RSID VISITNO ... HMP_BODY_SUBSITE SRS_SAMPLE_IDThis can also be converted to phyloseq for ecological and differential abundance analysis; see the HMP16SData vignette for details.
Bibliography
See individual function and methods documentation for specific details.↩︎
https://confluence.broadinstitute.org/display/GDAC/FAQ#FAQ-Q%C2%A0Whatreferencegenomebuildareyouusing↩︎
https://gdc.cancer.gov/about-data/data-harmonization-and-generation/genomic-data-harmonization-0↩︎
https://confluence.broadinstitute.org/display/GDAC/FAQ#FAQ-Q%C2%A0Whatreferencegenomebuildareyouusing↩︎
https://bioconductor.org/packages/release/bioc/vignettes/TCGAbiolinks/inst/doc/download_prepare.html↩︎