MultiAssayExperiment: Integrative workflow for public multi-omics data
Marcel Ramos
Roswell Park Comprehensive Cancer Center, Buffalo, NYLudwig Geistlinger
CUNY School of Public Health, New York, NYLevi Waldron
CUNY School of Public Health, New York, NYJuly 02, 2025
Source:vignettes/Ramos_MultiAssayExperiment.Rmd
Ramos_MultiAssayExperiment.RmdMulti-omic Integration and Analysis of cBioPortal and TCGA data with MultiAssayExperiment
Workshop participation
- See the instruction Google slides
- Browse to the workshop website. This page includes:
- Run this workshop on the free Orchestra Cloud platform (search for title: “Multi-omic Integration of cBioPortal and TCGA data with MultiAssayExperiment”)
- Docker users can run this workshop locally via:
docker run -e PASSWORD=bioc -p 8787:8787 ghcr.io/waldronlab/multiassayworkshop:latestRequirements: R/Bioconductor packages
The workshop uses a Docker container with Bioconductor version 3.21. If you would like to install Bioconductor on your computer at a later date, see the Bioconductor installation instructions.
Here is a list of packages that we will be using:
library(MultiAssayExperiment)
library(RaggedExperiment)
library(curatedTCGAData)
library(cBioPortalData)
library(TCGAutils)
library(SingleCellMultiModal)
library(UpSetR)
library(GenomicDataCommons)
library(GenomeInfoDb)Citing MultiAssayExperiment
Please use these citations (Ramos et al.
2017), (Ramos et al. 2020), and
(Ramos et al. 2023) when using
MultiAssayExperiment, curatedTCGAData,
cBioPortalData, or RaggedExperiment. Your
citations are very much appreciated!
Overview of Packages
| Package | Description |
|---|---|
| MultiAssayExperiment | Infrastructure package to represent multi-omics data |
| curatedTCGAData | Downloads TCGA data from ExperimentHub in MultiAssayExperiment form |
| terraTCGAdata | Obtains TCGA data within Terra cloud service as MultiAssayExperiment |
| cBioPortalData | Access over 300 study datasets from the cBio Genomics Portal |
| TCGAutils | Make use of utility functions for working with TCGA data |
| SingleCellMultiModal | Obtain single cell data from various multi-modality studies |
| TENxIO | Import 10X files as Bioconductor data classes |
Key Packages
MultiAssayExperiment
- provides an integrative representation for multi-omics data
- modelled after the
SummarizedExperimentrepresentation for expression data - easy-to-use operations for manipulating multiple sets of data such as copy number alterations, mutations, proteomics, methylation, and more
MultiAssayExperiment object schematic
cBioPortalData
- R/Bioconductor interface to cBioPortal data
- makes use of the revamped API with caching
- queries are handled for the user in the background
- easy-to-use interface (no knowledge of the cBioPortal data model required)
- see
getStudies()for a list of available studies
curatedTCGAData
- Many tools exist for accessing and downloading The
Cancer Genome Atlas data:
RTCGAToolbox,GenomicDataCommons,TCGAbiolinks,cBioPortalwebsite, Broad GDAC Firehose, and more - makes it easy to obtain user-friendly and integrative data at very little cognitive overhead
- conveniently places data in the analysis platform of choice, R/Bioconductor
- provides 33 different cancer types from the Broad GDAC Firehose
- On-the-fly construction from ‘flat’ files
-
hg19data -
MultiAssayExperimentrepresentations
Available Studies– (curatedTCGAData section) A list of available cancer studies fromTCGAutils::diseaseCodes.OmicsTypes– A descriptive table of ’omics types incuratedTCGAData(thanks to Ludwig G.@lgeistlinger)
terraTCGAdata
- Tool for making use of TCGA data within the Terra platform.
- Go to https://app.terra.bio and sign in with your Google account (credit card required)
- Select the latest RStudio / Bioconductor instance and install the package.
- Make sure to authenticate Google Cloud and check
AnVIL::gcloud_exists() -
findTCGAworkspacesto list all the available workspaces with TCGA data -
terraTCGAworkspaceto set the TCGA dataset to download -
terraTCGAdata- main function to download data
SingleCellMultiModal
- Serves multi-modal datasets from GEO and other sources as
MultiAssayExperimentdata representations. Some representations are out of memory using theHDF5format as well as theMTXformat. - Available technologies are scNMT, 10X Multiome, seqFISH, (EC)CITEseq, SCoPE2 and others.
TENxIO
- Allows import of 10X files as Bioconductor objects, e.g.,
TENxH5will produce aSingleCellExperimentfrom an.h5file. - Available for a number of formats such as
MTX,TSV, peaks, fragments, and.tar.gzfiles.
TCGAutils
- allows additional exploration, and manipulation of samples and metadata
- User-friendly operations for subsetting, separating, converting, and reshaping of sample and feature TCGA data
- developed specifically for TCGA data and
curatedTCGADataoutputs
It provides convenience / helper functions in three major areas:
- conversion / summarization of row annotations to genomic ranges
- identification and separation of samples
- translation and interpretation of TCGA identifiers
For the cheatsheet reference table, see the TCGAutils Cheatsheet.
To better understand how it all fits together, this schematic shows the relationship among all as part of the curatedTCGAData pipeline.
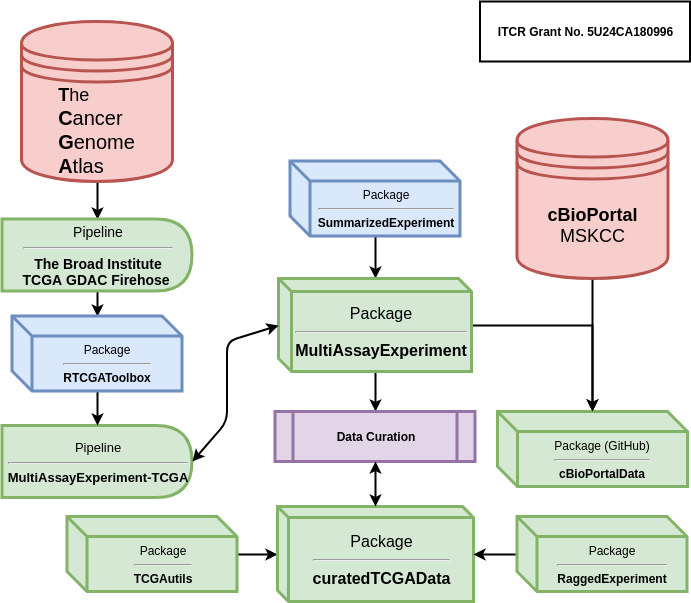
Schematic of curatedTCGAData Pipeline
Data Classes
This section summarizes three fundamental data classes for the representation of multi-omics experiments.
(Ranged)SummarizedExperiment
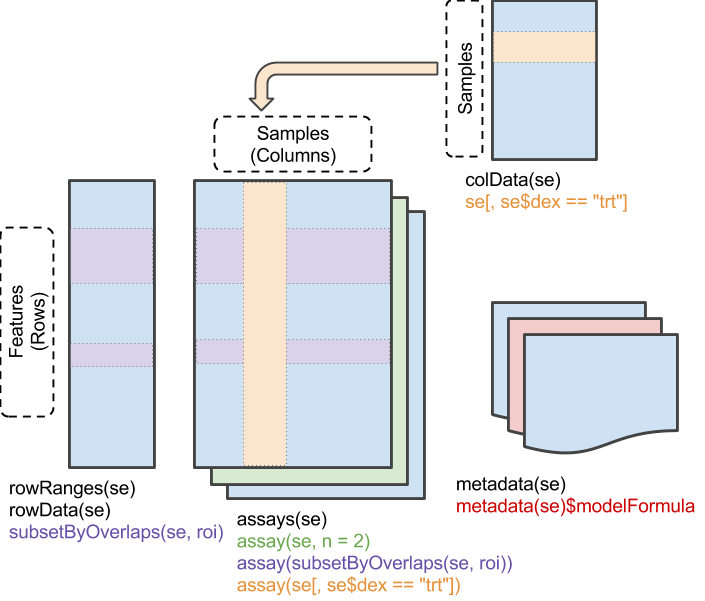
A matrix-like container where rows represent features of interest and columns represent samples. The objects contain one or more assays, each represented by a matrix-like object of numeric or other mode.
- matrix-like representation of experimental data including RNA sequencing and microarray experiments.
- stores multiple experimental data matrices of identical dimensions,
with associated metadata on:
- the rows/genes/transcripts/other measurements
(
rowData) - column/sample phenotype or clinical data (
colData) - overall experiment (
metadata).
- the rows/genes/transcripts/other measurements
(
-
RangedSummarizedExperimentassociates aGRangesorGRangesListvector with the rows
Note. Many other classes for experimental data are actually
derived from SummarizedExperiment (e.g.,
SingleCellExperiment for single-cell RNA sequencing
experiments)
## [1] "SingleCellExperiment" "RangedSummarizedExperiment"
## [3] "SummarizedExperiment" "RectangularData"
## [5] "Vector" "Annotated"
## [7] "vector_OR_Vector"
RaggedExperiment
- flexible representation for segmented copy number, somatic mutations
such as represented in
.vcffiles, and other ragged array schema for genomic location data. - similar to the
GRangesListclass inGenomicRanges - used to represent differing genomic ranges on each of a set of samples
showClass("RaggedExperiment")## Class "RaggedExperiment" [package "RaggedExperiment"]
##
## Slots:
##
## Name: assays rowidx colidx metadata
## Class: GRangesList integer integer list
##
## Extends: "Annotated"RaggedExperiment provides a flexible set of _*Assay_
methods to support transformation of ranged list data to matrix
format.
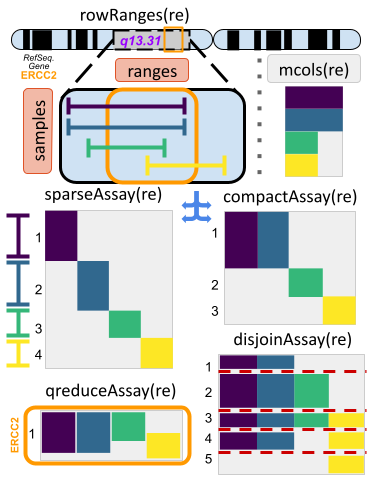
RaggedExperiment object schematic. Rows and columns represent genomic ranges and samples, respectively. Assay operations can be performed with (from left to right) compactAssay, qreduceAssay, and sparseAssay.
The Integrative Container
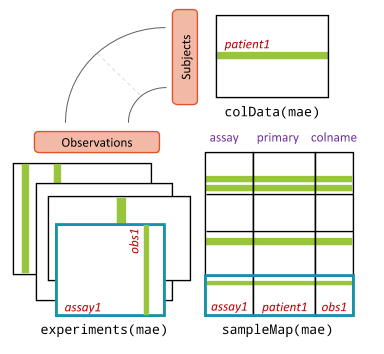
MultiAssayExperiment object schematic. colData provides data about the patients, cell lines, or other biological units, with one row per unit and one column per variable. The experiments are a list of assay datasets of arbitrary class. The sampleMap relates each column (observation) in ExperimentList to exactly one row (biological unit) in colData; however, one row of colData may map to zero, one, or more columns per assay, allowing for missing and replicate assays. sampleMap allows for per-assay sample naming conventions. Metadata can be used to store information in arbitrary format about the MultiAssayExperiment. Green stripes indicate a mapping of one subject to multiple observations across experiments.
MultiAssayExperiment
- coordinates multi-omics experiment data on a set of biological specimens
- can contain any number of assays with different representations and dimensions
- assays can be ID-based, where measurements are indexed identifiers of genes, microRNA, proteins, microbes, etc.
- assays may be range-based, where measurements correspond to
genomic ranges that can be represented as
GRangesobjects, such as gene expression or copy number.
Click on the fold to see what data classes are supported!
-
matrix: the most basic class for ID-based datasets, could be used for example for gene expression summarized per-gene, microRNA, metabolomics, or microbiome data. -
SummarizedExperimentand derived methods: described above, could be used for miRNA, gene expression, proteomics, or any matrix-like data where measurements are represented by IDs. -
RangedSummarizedExperiment: described above, could be used for gene expression, methylation, or other data types referring to genomic positions. -
ExpressionSet: Another rich representation for ID-based datasets, supported only for legacy reasons -
RaggedExperiment: described above, for non-rectangular (ragged) ranged-based datasets such as segmented copy number, where segmentation of copy number alterations occurs and different genomic locations in each sample. -
RangedVcfStack: For VCF archives broken up by chromosome (seeVcfStackclass defined in theGenomicFilespackage) -
DelayedMatrix: An on-disk representation of matrix-like objects for large datasets. It reduces memory usage and optimizes performance with delayed operations. This class is part of theDelayedArraypackage. -
SingleCellExperiment,SpatialExperiment, etc.
Note. Data classes that support row and column naming and
subsetting may be used in a MultiAssayExperiment.
MatchedAssayExperiment
- uniform subclass of
MultiAssayExperiment - “all patients have a sample in each assay”
# coercion
as(x, "MatchedAssayExperiment")
# construction from MAE
MatchedAssayExperiment(mae)Note. The MultiAssayExperiment package provides
functionality to merge replicate profiles for a single patient
(mergeReplicates()).
Key points
-
MultiAssayExperimentcoordinates different Bioconductor classes into one unified object -
MultiAssayExperimentis an infrastructure package whilecuratedTCGADataandcBioPortalDataprovide data on cancer studies including TCGA
Building from Scratch: MultiAssayExperiment
miniACC Demo
Get started by trying out MultiAssayExperiment using a
subset of the TCGA adrenocortical carcinoma (ACC) dataset provided with
the package. This dataset provides five assays on 92 patients, although
all five assays were not performed for every patient:
- RNASeq2GeneNorm: gene mRNA abundance by RNA-seq
- gistict: GISTIC genomic copy number by gene
- RPPAArray: protein abundance by Reverse Phase Protein Array
- Mutations: non-silent somatic mutations by gene
- miRNASeqGene: microRNA abundance by microRNA-seq.
data("miniACC")
miniACC## A MultiAssayExperiment object of 5 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 5:
## [1] RNASeq2GeneNorm: SummarizedExperiment with 198 rows and 79 columns
## [2] gistict: SummarizedExperiment with 198 rows and 90 columns
## [3] RPPAArray: SummarizedExperiment with 33 rows and 46 columns
## [4] Mutations: matrix with 97 rows and 90 columns
## [5] miRNASeqGene: SummarizedExperiment with 471 rows and 80 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat files
shiny Demo
Click Here to
open the shiny tutorial.
Key points
- Extractor functions allow users to take components from the
MultiAssayExperimentobject - They’re usually the same name as the component except for
experimentswhich extracts theExperimentList
Notes on Working with MultiAssayExperiment
API cheat sheet
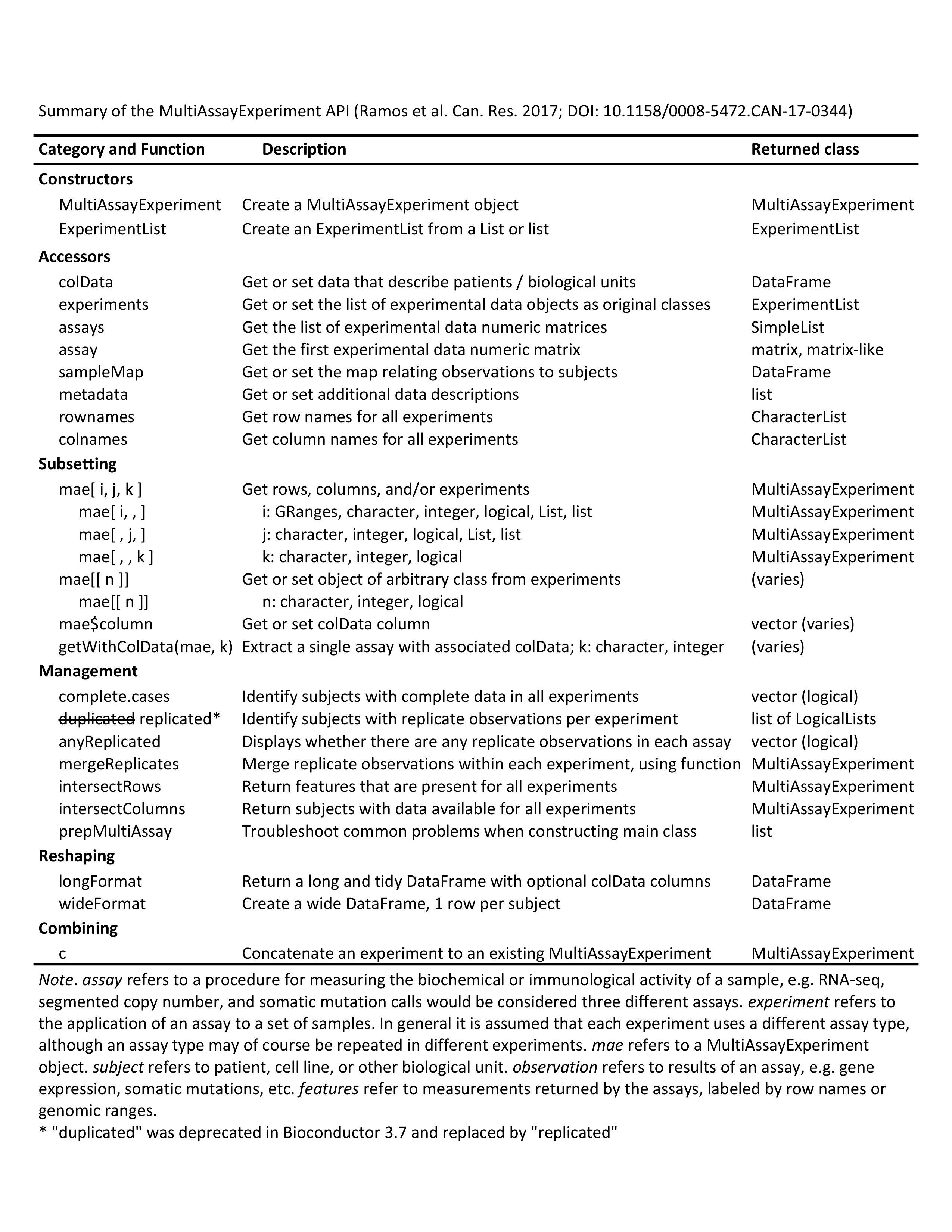
The MultiAssayExperiment API for construction, access, subsetting, management, and reshaping to formats for application of R/Bioconductor graphics and analysis packages.
MultiAssayExperiment construction and concatenation
constructor function
The MultiAssayExperiment constructor function accepts
three arguments:
-
experiments- AnExperimentListorlistof rectangular data -
colData- ADataFramedescribing the patients (or cell lines, or other biological units) -
sampleMap- ADataFrameofassay,primary, andcolnameidentifiers
The miniACC object can be reconstructed as follows:
MultiAssayExperiment(
experiments = experiments(miniACC),
colData = colData(miniACC),
sampleMap = sampleMap(miniACC),
metadata = metadata(miniACC)
)## A MultiAssayExperiment object of 5 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 5:
## [1] RNASeq2GeneNorm: SummarizedExperiment with 198 rows and 79 columns
## [2] gistict: SummarizedExperiment with 198 rows and 90 columns
## [3] RPPAArray: SummarizedExperiment with 33 rows and 46 columns
## [4] Mutations: matrix with 97 rows and 90 columns
## [5] miRNASeqGene: SummarizedExperiment with 471 rows and 80 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesGetting help with prepMultiAssay
The prepMultiAssay function allows the user to diagnose
typical problems when creating a MultiAssayExperiment
object. See ?prepMultiAssay for more details.
Combining MultiAssayExperiment object with the c
function
The c function allows the user to concatenate an
additional experiment to an existing MultiAssayExperiment.
The optional sampleMap argument allows concatenating an
assay whose column names do not match the row names of
colData. For convenience, the mapFrom argument
allows the user to map from a particular experiment
provided that the order of the
colnames is in the same. A warning will be
issued to make the user aware of this assumption. For example, to
concatenate a matrix of log2-transformed RNA-seq results:
## Warning: Assuming column order in the data provided
## matches the order in 'mapFrom' experiment(s) colnames
experiments(miniACC2)## ExperimentList class object of length 6:
## [1] RNASeq2GeneNorm: SummarizedExperiment with 198 rows and 79 columns
## [2] gistict: SummarizedExperiment with 198 rows and 90 columns
## [3] RPPAArray: SummarizedExperiment with 33 rows and 46 columns
## [4] Mutations: matrix with 97 rows and 90 columns
## [5] miRNASeqGene: SummarizedExperiment with 471 rows and 80 columns
## [6] log2rnaseq: matrix with 198 rows and 79 columnscolData - information biological units
This slot is a DataFrame describing the characteristics
of biological units, for example clinical data for patients. In the
prepared datasets from The
Cancer Genome Atlas, each row is one patient and each column is a
clinical, pathological, subtype, or other variable. The $
function provides a shortcut for accessing or setting
colData columns.
colData(miniACC)[1:4, 1:4]## DataFrame with 4 rows and 4 columns
## patientID years_to_birth vital_status days_to_death
## <character> <integer> <integer> <integer>
## TCGA-OR-A5J1 TCGA-OR-A5J1 58 1 1355
## TCGA-OR-A5J2 TCGA-OR-A5J2 44 1 1677
## TCGA-OR-A5J3 TCGA-OR-A5J3 23 0 NA
## TCGA-OR-A5J4 TCGA-OR-A5J4 23 1 423
table(miniACC$race)##
## asian black or african american white
## 2 1 78Note. MultiAssayExperiment supports both missing
observations and replicate observations, i.e., one row of
colData can map to 0, 1, or more columns of any of the
experimental data matrices. One could therefore treat replicate
observations as one or multiple rows of colData. This can
result in different subsetting, duplicated(), and
wideFormat() behaviors.
Note. Multiple time points, or distinct biological replicates, can be
separate rows of the colData.
Key points
- Each row maps to zero or more observations in each experiment
- Usually, organized as one row per biological unit
ExperimentList - experiment data
Experimental datasets can be input as either a base list
or ExperimentList object for the set of samples collected.
To see the experiments use the experiments getter
function.
experiments(miniACC)## ExperimentList class object of length 5:
## [1] RNASeq2GeneNorm: SummarizedExperiment with 198 rows and 79 columns
## [2] gistict: SummarizedExperiment with 198 rows and 90 columns
## [3] RPPAArray: SummarizedExperiment with 33 rows and 46 columns
## [4] Mutations: matrix with 97 rows and 90 columns
## [5] miRNASeqGene: SummarizedExperiment with 471 rows and 80 columnsNote. Each matrix column must correspond to exactly one row in
colData. In other words, each patient or cell line must be
traceable. However, multiple columns can come from the same patient, or
there can be no data for that patient.
Key points:
- One rectangular dataset per list element
- One column per assayed specimen.
- Matrix rows correspond to variables, e.g. genes or genomic ranges
-
ExperimentListelements can be genomic range-based (e.g.SummarizedExperimentorRaggedExperiment) or ID-based data - Most rectangular-type data classes are supported
Note. Any data class can be included in the
ExperimentList, as long as it supports: single-bracket
subsetting ([), dimnames, and
dim. Most data classes defined in Bioconductor meet these
requirements.
sampleMap - relationship graph
sampleMap is a graph representation of the relationship
between biological units and experimental results. In simple cases where
the column names of ExperimentList data matrices match the
row names of colData, the user won’t need to specify or
think about a sample map, it can be created automatically by the
MultiAssayExperiment constructor. sampleMap is
a simple three-column DataFrame:
-
assaycolumn: the name of the assay, and found in the names ofExperimentListlist names -
primarycolumn: identifiers of patients or biological units, and found in the row names ofcolData -
colnamecolumn: identifiers of assay results, and found in the column names ofExperimentListelements Helper functions are available for creating a map from a list. See?listToMap
sampleMap(miniACC)## DataFrame with 385 rows and 3 columns
## assay primary colname
## <factor> <character> <character>
## 1 RNASeq2GeneNorm TCGA-OR-A5J1 TCGA-OR-A5J1-01A-11R..
## 2 RNASeq2GeneNorm TCGA-OR-A5J2 TCGA-OR-A5J2-01A-11R..
## 3 RNASeq2GeneNorm TCGA-OR-A5J3 TCGA-OR-A5J3-01A-11R..
## 4 RNASeq2GeneNorm TCGA-OR-A5J5 TCGA-OR-A5J5-01A-11R..
## 5 RNASeq2GeneNorm TCGA-OR-A5J6 TCGA-OR-A5J6-01A-31R..
## ... ... ... ...
## 381 miRNASeqGene TCGA-PA-A5YG TCGA-PA-A5YG-01A-11R..
## 382 miRNASeqGene TCGA-PK-A5H8 TCGA-PK-A5H8-01A-11R..
## 383 miRNASeqGene TCGA-PK-A5H9 TCGA-PK-A5H9-01A-11R..
## 384 miRNASeqGene TCGA-PK-A5HA TCGA-PK-A5HA-01A-11R..
## 385 miRNASeqGene TCGA-PK-A5HB TCGA-PK-A5HB-01A-11R..Key points:
- relates experimental observations (
colnames) tocolData - permits experiment-specific sample naming, missing, and replicate observations
metadata
Metadata can be used to keep additional information about patients,
assays performed on individuals or on the entire cohort, or features
such as genes, proteins, and genomic ranges. There are many options
available for storing metadata. First, MultiAssayExperiment
has its own metadata for describing the entire experiment:
metadata(miniACC)## $title
## [1] "Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma"
##
## $PMID
## [1] "27165744"
##
## $sourceURL
## [1] "http://s3.amazonaws.com/multiassayexperiments/accMAEO.rds"
##
## $RPPAfeatureDataURL
## [1] "http://genomeportal.stanford.edu/pan-tcga/show_target_selection_file?filename=Allprotein.txt"
##
## $colDataExtrasURL
## [1] "http://www.cell.com/cms/attachment/2062093088/2063584534/mmc3.xlsx"Additionally, the DataFrame class used by
sampleMap and colData, as well as the
ExperimentList class, similarly support metadata. Finally,
many experimental data objects that can be used in the
ExperimentList support metadata. These provide flexible
options to users and to developers of derived classes.
The Cancer Genome Atlas (TCGA) Data from
curatedTCGAData
Most unrestricted TCGA data (from 33 cancer types) are available as
MultiAssayExperiment objects from the
curatedTCGAData package. This represents a lot of
harmonization!
Here we list the available data for the Adrenocortical Carcinoma
("ACC") cancer type:
library(curatedTCGAData)
curatedTCGAData("ACC", version = "2.0.1", dry.run = TRUE)## ah_id title file_size
## 1 EH4737 ACC_CNASNP-20160128 0.8 Mb
## 2 EH4738 ACC_CNVSNP-20160128 0.2 Mb
## 3 EH4740 ACC_GISTIC_AllByGene-20160128 0.2 Mb
## 4 EH4741 ACC_GISTIC_Peaks-20160128 0 Mb
## 5 EH4742 ACC_GISTIC_ThresholdedByGene-20160128 0.2 Mb
## 6 EH4744 ACC_Methylation-20160128_assays 239.2 Mb
## 7 EH4745 ACC_Methylation-20160128_se 6 Mb
## 8 EH4746 ACC_miRNASeqGene-20160128 0.1 Mb
## 9 EH4747 ACC_Mutation-20160128 0.7 Mb
## 10 EH4748 ACC_RNASeq2Gene-20160128 2.7 Mb
## 11 EH4749 ACC_RNASeq2GeneNorm-20160128 4 Mb
## 12 EH4750 ACC_RPPAArray-20160128 0.1 Mb
## rdataclass rdatadateadded rdatadateremoved
## 1 RaggedExperiment 2021-01-27 <NA>
## 2 RaggedExperiment 2021-01-27 <NA>
## 3 SummarizedExperiment 2021-01-27 <NA>
## 4 RangedSummarizedExperiment 2021-01-27 <NA>
## 5 SummarizedExperiment 2021-01-27 <NA>
## 6 SummarizedExperiment 2021-01-27 <NA>
## 7 RaggedExperiment 2021-01-27 <NA>
## 8 SummarizedExperiment 2021-01-27 <NA>
## 9 SummarizedExperiment 2021-01-27 <NA>
## 10 SummarizedExperiment 2021-01-27 <NA>
## 11 DFrame 2021-01-27 <NA>
## 12 SummarizedExperiment 2021-01-27 <NA>We then download the data with dry.run set to
FALSE.
acc <- curatedTCGAData(
diseaseCode = "ACC",
assays = c(
"miRNASeqGene", "RPPAArray", "Mutation", "RNASeq2GeneNorm", "CNVSNP"
),
version = "2.0.1",
dry.run = FALSE
)
acc## A MultiAssayExperiment object of 5 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 5:
## [1] ACC_CNVSNP-20160128: RaggedExperiment with 21052 rows and 180 columns
## [2] ACC_miRNASeqGene-20160128: SummarizedExperiment with 1046 rows and 80 columns
## [3] ACC_Mutation-20160128: RaggedExperiment with 20166 rows and 90 columns
## [4] ACC_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 79 columns
## [5] ACC_RPPAArray-20160128: SummarizedExperiment with 192 rows and 46 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesThese objects contain most unrestricted TCGA assay and clinical / pathological data, as well as some curated data from the supplements of published TCGA primary papers at the end of the colData columns:
## [1] 92 822## [1] "MethyLevel" "miRNA.cluster" "SCNA.cluster" "protein.cluster"
## [5] "COC" "OncoSign" "purity" "ploidy"
## [9] "genome_doublings" "ADS"
cBioPortalData
The cBioPortal for Cancer Genomics provides access to hundreds of datasets collected and curated from different institutions. cBioPortalData is an R/Bioconductor client to download these datasets and integrate them using the MultiAssayExperiment data class. There is a more detailed workshop dedicated to cBioPortalData.
There are two main ways of accessing this data using the cBioPortalData package:
-
cBioDataPack- tarball (.tar.gz) data files -
cBioPortalData- data from the API
Note. pkgdown reference website here: https://waldronlab.io/cBioPortalData/
Listing the studies available
First, we create an API object using the cBioPortal
function. This will allow us to subsequently generate queries for the
service.
cbio <- cBioPortal()This function can also access local and private cBioPortal servers;
see ?cBioPortal.
Setting the buildReport argument to the
getStudies function adds two columns indicating whether the
study can be accessed via API (for accessing data only for specific
genes, gene panels, or molecular assays), and via data-pack (for
accessing all available data).
getStudies(cbio, buildReport = TRUE)## # A tibble: 494 × 15
## name description publicStudy pmid citation groups status importDate
## <chr> <chr> <lgl> <chr> <chr> <chr> <int> <chr>
## 1 Acute Lympho… Comprehens… TRUE 2573… Anderss… "PUBL… 0 2024-12-0…
## 2 Hypodiploid … Whole geno… TRUE 2333… Holmfel… "" 0 2024-12-0…
## 3 Adenoid Cyst… Targeted S… TRUE 2441… Ross et… "ACYC… 0 2024-12-0…
## 4 Adenoid Cyst… Whole-geno… TRUE 2686… Rettig … "ACYC… 0 2024-12-0…
## 5 Adenoid Cyst… WGS of 21 … TRUE 2663… Mitani … "ACYC… 0 2024-12-0…
## 6 Adenoid Cyst… Whole-geno… TRUE 2682… Drier e… "ACYC" 0 2024-12-0…
## 7 Adenoid Cyst… Whole exom… TRUE 2377… Stephen… "ACYC… 0 2024-12-0…
## 8 Acute Lympho… Whole-geno… TRUE 2777… Zhang e… "PUBL… 0 2024-12-0…
## 9 Appendiceal … Targeted s… TRUE 3649… Michael… "PUBL… 0 2024-12-0…
## 10 Bladder Canc… Whole exom… TRUE 2690… Al-Ahma… "" 0 2024-12-0…
## # ℹ 484 more rows
## # ℹ 7 more variables: allSampleCount <int>, readPermission <lgl>,
## # studyId <chr>, cancerTypeId <chr>, referenceGenome <chr>, api_build <lgl>,
## # pack_build <lgl>This adds two additional columns to the end of the dataset reporting
the status of the builds. Not all studies can be converted to
MultiAssayExperiment. Some studies require additional
cleaning to be represented with MultiAssayExperiment.
cBioDataPack
The cBioDataPack function downloads complete datasets
and integrates them as a MultiAssayExperiment object. The
names.field argument allows changing how GenomicRanges are
labelled (HUGO, Entrez, Gene Names). There are other arguments but
you’re unlikely to use them.
library(cBioPortalData)
(uvm <- cBioDataPack("uvm_tcga"))## A MultiAssayExperiment object of 8 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 8:
## [1] cna_hg19.seg: RaggedExperiment with 7618 rows and 80 columns
## [2] cna: SummarizedExperiment with 24776 rows and 80 columns
## [3] linear_cna: SummarizedExperiment with 24776 rows and 80 columns
## [4] methylation_hm450: SummarizedExperiment with 15468 rows and 80 columns
## [5] mrna_seq_v2_rsem_zscores_ref_all_samples: SummarizedExperiment with 20531 rows and 80 columns
## [6] mrna_seq_v2_rsem_zscores_ref_diploid_samples: SummarizedExperiment with 20440 rows and 80 columns
## [7] mrna_seq_v2_rsem: SummarizedExperiment with 20531 rows and 80 columns
## [8] mutations: RaggedExperiment with 2174 rows and 80 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filescBioPortalData
This function accesses data from the cBioPortal API. It requires
specifying which of the available molecular profiles for a given study
to fetch. For example, the urcc_mskcc_2016 study has
molecular profiles:
(mp <- molecularProfiles(cbio, studyId = "urcc_mskcc_2016"))## # A tibble: 2 × 8
## molecularAlterationType datatype name description showProfileInAnalysi…¹
## <chr> <chr> <chr> <chr> <lgl>
## 1 COPY_NUMBER_ALTERATION DISCRETE Putative … Putative c… TRUE
## 2 MUTATION_EXTENDED MAF Mutations Mutation s… TRUE
## # ℹ abbreviated name: ¹showProfileInAnalysisTab
## # ℹ 3 more variables: patientLevel <lgl>, molecularProfileId <chr>,
## # studyId <chr>
mp[["molecularProfileId"]]## [1] "urcc_mskcc_2016_cna" "urcc_mskcc_2016_mutations"It also requires specifying a subset of genes, using either ‘genes’ with a vector of Entrez IDs, or ‘genePanelId’ with a gene panel ID:
genePanels(cbio)## # A tibble: 66 × 2
## description genePanelId
## <chr> <chr>
## 1 Targeted (27 cancer genes) sequencing of adenoid cystic carcinom… ACYC_FMI_27
## 2 Targeted panel of 232 genes. Agilent
## 3 Targeted panel of 8 genes. AmpliSeq
## 4 ARCHER-HEME gene panel (199 genes) ARCHER-HEM…
## 5 ARCHER-SOLID Gene Panel (62 genes) ARCHER-SOL…
## 6 Targeted sequencing of various tumor types via bait v3. bait_v3
## 7 Targeted sequencing of various tumor types via bait v4. bait_v4
## 8 Targeted sequencing of various tumor types via bait v5. bait_v5
## 9 Targeted panel of 387 cancer-related genes. bcc_unige_…
## 10 Research (CMO) IMPACT-Heme gene panel version 3. HemePACT_v3
## # ℹ 56 more rows
getGenePanel(cbio, genePanelId= "IMPACT230")## # A tibble: 230 × 2
## entrezGeneId hugoGeneSymbol
## <int> <chr>
## 1 25 ABL1
## 2 27 ABL2
## 3 207 AKT1
## 4 208 AKT2
## 5 10000 AKT3
## 6 238 ALK
## 7 242 ALOX12B
## 8 139285 AMER1
## 9 324 APC
## 10 367 AR
## # ℹ 220 more rowsThe getGenePanel option only really makes sense for
studies that employed targeted sequencing of a gene panel, or that
employed whole-exome or whole-genome assays, if you are content with
only the subset of genes in the panel. This argument will subset
available genes to those in the panel, whether or not that panel was
used in the study. If you want all available data, use the
cBioDataPack function instead.
From the (Chen et al. 2016) publication
of the [urcc_mskcc_2016][] study, we know that the study used the
IMPACT230 gene panel, so we will specify that.
urcc <- cBioPortalData(
api = cbio,
studyId = "urcc_mskcc_2016",
genePanelId = "IMPACT230",
molecularProfileIds =
c("urcc_mskcc_2016_cna", "urcc_mskcc_2016_mutations")
)## harmonizing input:
## removing 8 colData rownames not in sampleMap 'primary'
urcc## A MultiAssayExperiment object of 2 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 2:
## [1] urcc_mskcc_2016_mutations: RangedSummarizedExperiment with 156 rows and 53 columns
## [2] urcc_mskcc_2016_cna: SummarizedExperiment with 31 rows and 8 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesKey points
-
curatedTCGADataprovides TCGA data with some curation including tumor subtype information -
cBioPortalDatahas two main functions, one for downloading pre-packaged data and another for sending queries through the cBioPortal API
Utilities for TCGA
Aside from the available reshaping functions already included in the
MultiAssayExperiment package, the TCGAutils
package provides additional helper functions for working with TCGA
data.
A number of helper functions are available for managing datasets from
curatedTCGAData. These include:
- Conversions of
SummarizedExperimenttoRangedSummarizedExperimentbased onTxDb.Hsapiens.UCSC.hg19.knownGenefor:-
symbolsToRanges(): gene symbols -
qreduceTCGA(): convertRaggedExperimentobjects toRangedSummarizedExperimentwith one row per gene symbol, for:- segmented copy number datasets (“CNVSNP” and “CNASNP”)
- somatic mutation datasets (“Mutation”), with a value of 1 for any non-silent mutation and a value of 0 for no mutation or silent mutation
-
(1) Conversion of row metadata for curatedTCGAData
objects
qreduceTCGA
The qreduceTCGA function converts
RaggedExperiment mutation data objects to
RangedSummarizedExperiment using org.Hs.eg.db
and the qreduceTCGA utility function from
RaggedExperiment to summarize ‘silent’ and ‘non-silent’
mutations based on a ‘Variant_Classification’ metadata column in the
original object.
## Update build metadata to "hg19"
genome(acc[["ACC_Mutation-20160128"]]) <- "NCBI37"
seqlevelsStyle(acc[["ACC_Mutation-20160128"]]) <- "UCSC"
gnome <- genome(acc[["ACC_Mutation-20160128"]])
gnome <- translateBuild(gnome)
genome(acc[["ACC_Mutation-20160128"]]) <- gnome
qreduceTCGA(acc)## ## 403 genes were dropped because they have exons located on both strands of the
## same reference sequence or on more than one reference sequence, so cannot be
## represented by a single genomic range.
## Use 'single.strand.genes.only=FALSE' to get all the genes in a GRangesList
## object, or use suppressMessages() to suppress this message.## Warning in (function (seqlevels, genome, new_style) : cannot switch some hg19's
## seqlevels from UCSC to NCBI style## 'select()' returned 1:1 mapping between keys and columns## Warning in .normarg_seqlevelsStyle(value): more than one seqlevels style
## supplied, using the 1st one only
## Warning in .normarg_seqlevelsStyle(value): cannot switch some hg19's seqlevels
## from UCSC to NCBI style## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 270 sampleMap rows not in names(experiments)## A MultiAssayExperiment object of 5 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 5:
## [1] ACC_miRNASeqGene-20160128: SummarizedExperiment with 1046 rows and 80 columns
## [2] ACC_RNASeq2GeneNorm-20160128: SummarizedExperiment with 20501 rows and 79 columns
## [3] ACC_RPPAArray-20160128: SummarizedExperiment with 192 rows and 46 columns
## [4] ACC_Mutation-20160128_simplified: RangedSummarizedExperiment with 22912 rows and 90 columns
## [5] ACC_CNVSNP-20160128_simplified: RangedSummarizedExperiment with 22912 rows and 180 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat files
symbolsToRanges
In the cases where row annotations indicate gene symbols, the
symbolsToRanges utility function converts genes to genomic
ranges and replaces existing assays with
RangedSummarizedExperiment objects. Gene annotations are
given as ‘hg19’ genomic regions.
symbolsToRanges(acc)## 403 genes were dropped because they have exons located on both strands of the
## same reference sequence or on more than one reference sequence, so cannot be
## represented by a single genomic range.
## Use 'single.strand.genes.only=FALSE' to get all the genes in a GRangesList
## object, or use suppressMessages() to suppress this message.## Warning in (function (seqlevels, genome, new_style) : cannot switch some hg19's
## seqlevels from UCSC to NCBI style## 'select()' returned 1:1 mapping between keys and columns## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 79 sampleMap rows not in names(experiments)## A MultiAssayExperiment object of 6 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 6:
## [1] ACC_CNVSNP-20160128: RaggedExperiment with 21052 rows and 180 columns
## [2] ACC_miRNASeqGene-20160128: SummarizedExperiment with 1046 rows and 80 columns
## [3] ACC_Mutation-20160128: RaggedExperiment with 20166 rows and 90 columns
## [4] ACC_RPPAArray-20160128: SummarizedExperiment with 192 rows and 46 columns
## [5] ACC_RNASeq2GeneNorm-20160128_ranged: RangedSummarizedExperiment with 16954 rows and 79 columns
## [6] ACC_RNASeq2GeneNorm-20160128_unranged: SummarizedExperiment with 3547 rows and 79 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat files
simplifyTCGA
The simplifyTCGA function combines all of the above
operations to create a more managable MultiAssayExperiment
object and using RangedSummarizedExperiment assays where
possible.
TCGAutils::simplifyTCGA(acc)## 403 genes were dropped because they have exons located on both strands of the
## same reference sequence or on more than one reference sequence, so cannot be
## represented by a single genomic range.
## Use 'single.strand.genes.only=FALSE' to get all the genes in a GRangesList
## object, or use suppressMessages() to suppress this message.## Warning in (function (seqlevels, genome, new_style) : cannot switch some hg19's
## seqlevels from UCSC to NCBI style## 'select()' returned 1:1 mapping between keys and columns## Warning in .normarg_seqlevelsStyle(value): more than one seqlevels style
## supplied, using the 1st one only
## Warning in .normarg_seqlevelsStyle(value): cannot switch some hg19's seqlevels
## from UCSC to NCBI style## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 270 sampleMap rows not in names(experiments)## Warning in (function (seqlevels, genome, new_style) : cannot switch some hg19's
## seqlevels from UCSC to NCBI style## harmonizing input:
## removing 80 sampleMap rows not in names(experiments)
## 403 genes were dropped because they have exons located on both strands of the
## same reference sequence or on more than one reference sequence, so cannot be
## represented by a single genomic range.
## Use 'single.strand.genes.only=FALSE' to get all the genes in a GRangesList
## object, or use suppressMessages() to suppress this message.## Warning in (function (seqlevels, genome, new_style) : cannot switch some hg19's
## seqlevels from UCSC to NCBI style## 'select()' returned 1:1 mapping between keys and columns## harmonizing input:
## removing 79 sampleMap rows not in names(experiments)## A MultiAssayExperiment object of 7 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 7:
## [1] ACC_RPPAArray-20160128: SummarizedExperiment with 192 rows and 46 columns
## [2] ACC_Mutation-20160128_simplified: RangedSummarizedExperiment with 22912 rows and 90 columns
## [3] ACC_CNVSNP-20160128_simplified: RangedSummarizedExperiment with 22912 rows and 180 columns
## [4] ACC_miRNASeqGene-20160128_ranged: RangedSummarizedExperiment with 1002 rows and 80 columns
## [5] ACC_miRNASeqGene-20160128_unranged: SummarizedExperiment with 44 rows and 80 columns
## [6] ACC_RNASeq2GeneNorm-20160128_ranged: RangedSummarizedExperiment with 16954 rows and 79 columns
## [7] ACC_RNASeq2GeneNorm-20160128_unranged: SummarizedExperiment with 3547 rows and 79 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesWhat types of samples are in the data?
Solution
The sampleTables function gives you an overview of
samples in each assay:
sampleTables(acc)## $`ACC_CNVSNP-20160128`
##
## 01 10 11
## 90 85 5
##
## $`ACC_miRNASeqGene-20160128`
##
## 01
## 80
##
## $`ACC_Mutation-20160128`
##
## 01
## 90
##
## $`ACC_RNASeq2GeneNorm-20160128`
##
## 01
## 79
##
## $`ACC_RPPAArray-20160128`
##
## 01
## 46Interpretation of sample codes:
## Code Definition Short.Letter.Code
## 1 01 Primary Solid Tumor TP
## 2 02 Recurrent Solid Tumor TR
## 3 03 Primary Blood Derived Cancer - Peripheral Blood TB
## 4 04 Recurrent Blood Derived Cancer - Bone Marrow TRBM
## 5 05 Additional - New Primary TAP
## 6 06 Metastatic TM
TCGAsplitAssays: separate the data from different
tissue types
TCGA datasets include multiple -omics for solid tumors, adjacent
normal tissues, blood-derived cancers and normals, and other tissue
types, which may be mixed together in a single dataset. The
MultiAssayExperiment object generated here has one patient
per row of its colData, but each patient may have two or
more -omics profiles by any assay, whether due to assaying of different
types of tissues or to technical replication.
TCGAsplitAssays separates profiles from different tissue
types (such as tumor and adjacent normal) into different assays of the
MultiAssayExperiment by taking a vector of sample codes,
and partitioning the current assays into assays with an appended sample
code:
split_acc <- TCGAsplitAssays(acc, c("01", "11"))## Warning: Some 'sampleCodes' not found in assays## Warning in .checkBarcodes(barcodes): Inconsistent barcode lengths: 28, 27Only about 43 participants have data across all experiments.
Curated molecular subtypes
Is there subtype data available in the
MultiAssayExperiment obtained from
curatedTCGAData?
Solution
The getSubtypeMap function will show actual variable
names found in colData that contain subtype information.
This can only be obtained from MultiAssayExperiment objects
provided by curatedTCGAData.
getSubtypeMap(acc)## ACC_annotations ACC_subtype
## 1 Patient_ID patientID
## 2 histological_subtypes Histology
## 3 mrna_subtypes C1A/C1B
## 4 mrna_subtypes mRNA_K4
## 5 cimp MethyLevel
## 6 microrna_subtypes miRNA cluster
## 7 scna_subtypes SCNA cluster
## 8 protein_subtypes protein cluster
## 9 integrative_subtypes COC
## 10 mutation_subtypes OncoSign## [1] "Usual Type" "Usual Type" "Usual Type" "Usual Type" "Usual Type"
## [6] "Usual Type"(3) Translation and Interpretation of TCGA identifiers
TCGAutils provides a number of ID translation functions.
These allow the user to translate from either file or case UUIDs to TCGA
barcodes and back. These functions work by querying the Genomic Data
Commons API via the GenomicDataCommons package (thanks to
Sean Davis). These include:
UUIDtoBarcode() - UUID to TCGA barcode
Here we have a known case UUID that we want to translate into a TCGA barcode.
UUIDtoBarcode("ae55b2d3-62a1-419e-9f9a-5ddfac356db4", from_type = "case_id")## case_id submitter_id
## 1 ae55b2d3-62a1-419e-9f9a-5ddfac356db4 TCGA-B0-5117In cases where we want to translate a known file UUID to the
associated TCGA patient barcode, we can use
UUIDtoBarcode.
UUIDtoBarcode("b4bce3ff-7fdc-4849-880b-56f2b348ceac", from_type = "file_id")## file_id associated_entities.entity_submitter_id
## 1 b4bce3ff-7fdc-4849-880b-56f2b348ceac TCGA-B0-5094-11A-01D-1421-08
barcodeToUUID() - TCGA barcode to UUID
Here we translate the first two TCGA barcodes of the previous copy-number alterations dataset to UUID:
## [1] "TCGA-OR-A5J1-01A-11D-A29H-01" "TCGA-OR-A5J1-10A-01D-A29K-01"
## [3] "TCGA-OR-A5J2-01A-11D-A29H-01" "TCGA-OR-A5J2-10A-01D-A29K-01"
barcodeToUUID(xbarcode)## submitter_aliquot_ids aliquot_ids
## 18 TCGA-OR-A5J1-01A-11D-A29H-01 1387b6c7-48fe-4961-86a7-0bdcbd3fef92
## 16 TCGA-OR-A5J1-10A-01D-A29K-01 cb537629-6a01-4d67-84ea-dbf130bd59c7
## 8 TCGA-OR-A5J2-01A-11D-A29H-01 6f0290b0-4cb4-4f72-853e-9ac363bd2c3b
## 11 TCGA-OR-A5J2-10A-01D-A29K-01 4bf2e4ac-399f-4a00-854b-8e23b561bb4d
UUIDtoUUID() - file and case IDs
We can also translate from file UUIDs to case UUIDs and vice versa as
long as we know the input type. We can use the case UUID from the
previous example to get the associated file UUIDs using
UUIDtoUUID. Note that this translation is a one to many
relationship, thus yielding a data.frame of file UUIDs for
a single case UUID.
head(UUIDtoUUID("ae55b2d3-62a1-419e-9f9a-5ddfac356db4", to_type = "file_id"))## case_id files.file_id
## 1 ae55b2d3-62a1-419e-9f9a-5ddfac356db4 472cecc2-4d28-45c8-be44-d7b1a140c549
## 2 ae55b2d3-62a1-419e-9f9a-5ddfac356db4 78f1f4b2-1ee9-4c8f-92a2-fe437eeba4c5
## 3 ae55b2d3-62a1-419e-9f9a-5ddfac356db4 ba07ad3d-5e92-4091-a853-5f9e49327378
## 4 ae55b2d3-62a1-419e-9f9a-5ddfac356db4 c0669db0-a20c-4836-aca7-0deb6ae8ba0c
## 5 ae55b2d3-62a1-419e-9f9a-5ddfac356db4 0ed8caf1-6bf8-4cf4-bc60-ac5b833418c0
## 6 ae55b2d3-62a1-419e-9f9a-5ddfac356db4 cb0f27bc-8403-414c-84a2-d08f26e3f464One possible way to verify that file IDs are matching case UUIDS is
to browse to the Genomic Data Commons webpage with the specific file
UUID. Here we look at the first file UUID entry in the output
data.frame:
https://portal.gdc.cancer.gov/files/0b4acc9e-3933-4d74-916a-a53c4a0665e6
In the page we check that the case UUID matches the input.
filenameToBarcode() - Using file names as input
fquery <- files() |>
GenomicDataCommons::filter(~ cases.project.project_id == "TCGA-ACC" &
data_category == "Copy Number Variation" &
data_type == "Copy Number Segment")
fnames <- head(results(fquery)$file_name)
filenameToBarcode(fnames)## file_name
## 1 GAMED_p_TCGA_B_312_313_314_NSP_GenomeWideSNP_6_E04_1361650.grch38.seg.v2.txt
## 2 6bea41f8-ad23-450e-b7ce-7c098db13640_wgs_gdc_realn.cr.igv.reheader.seg.txt
## 3 GAMED_p_TCGA_B_312_313_314_NSP_GenomeWideSNP_6_E05_1361692.grch38.seg.v2.txt
## 4 GAMED_p_TCGA_B_312_313_314_NSP_GenomeWideSNP_6_E06_1361784.grch38.seg.v2.txt
## 5 6101b5c5-c3d5-4f81-a721-dcdc5afdbffc_wgs_gdc_realn.cr.igv.reheader.seg.txt
## 6 GAMED_p_TCGA_B_312_313_314_NSP_GenomeWideSNP_6_D12_1361734.grch38.seg.v2.txt
## file_id
## 1 e88d7597-e2ea-41d3-a419-be70b5b49773
## 2 d2c5eef1-ea06-473b-b570-cce0486e4432
## 3 cf00f0a4-4a71-4b1b-a559-5ba7db28a184
## 4 5dfae645-caa6-4021-8f29-54e3cf043d33
## 5 73a58f81-388b-4723-8862-228b8db91020
## 6 63f7796d-85dc-4f02-85cb-983168afba66
## samples.portions.analytes.aliquots.submitter_id
## 1 TCGA-OR-A5KS-01A-11D-A309-01
## 2 TCGA-OR-A5KS-01A-11D-A75V-36
## 3 TCGA-OR-A5KS-10A-01D-A309-01
## 4 TCGA-OR-A5L1-01A-11D-A309-01
## 5 TCGA-OR-A5L1-10A-01D-A75V-36
## 6 TCGA-OR-A5KP-10A-01D-A309-01
## samples.portions.analytes.aliquots.submitter_id
## 1 TCGA-OR-A5KS-01A-11D-A309-01
## 2 TCGA-OR-A5KS-10A-01D-A75V-36
## 3 TCGA-OR-A5KS-10A-01D-A309-01
## 4 TCGA-OR-A5L1-01A-11D-A309-01
## 5 TCGA-OR-A5L1-01A-11D-A75V-36
## 6 TCGA-OR-A5KP-10A-01D-A309-01See the TCGAutils vignette page for more details.
Key points
-
TCGAutilsprovides users additional tools for modifying row and column metadata - The package works mainly with TCGA data including barcode identifiers
Data Management with MultiAssayExperiment
Single bracket [ subsetting
In the pseudo code below, the subsetting operations work on the rows of the following indices:
- i experimental data rows
-
j the primary names (vector input) or the column names
(
listorListinputs) - k assay
multiassayexperiment[i = rownames, j = primary or colnames, k = assay]Subsetting operations always return another
MultiAssayExperiment. For example, the following will
return any rows named “MAPK14” or “IGFBP2”, and remove any assays where
no rows match:
miniACC[c("MAPK14", "IGFBP2"), , ]The following will keep only patients of pathological stage IV, and all their associated assays:
stg4 <- miniACC$pathologic_stage == "stage iv"
# remove NA values from vector
miniACC[, stg4 & !is.na(stg4), ]And the following will keep only the RNA-seq dataset, and only patients for which this assay is available:
miniACC[, , "RNASeq2GeneNorm"]## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 306 sampleMap rows not in names(experiments)
## removing 13 colData rownames not in sampleMap 'primary'Subsetting by genomic ranges
If any ExperimentList objects have features represented by genomic
ranges (e.g. RangedSummarizedExperiment,
RaggedExperiment), then a GRanges object in
the first subsetting position will subset these objects as in
GenomicRanges::findOverlaps(). Any non-ranged
ExperimentList element will be subset to zero rows.
Double bracket [[ subsetting
The “double bracket” method ([[) is a convenience
function for extracting a single element of the
MultiAssayExperiment ExperimentList. It avoids
the use of experiments(mae)[[1L]]. For example, both of the
following extract the ExpressionSet object containing
RNA-seq data:
miniACC[[1L]]## class: SummarizedExperiment
## dim: 198 79
## metadata(3): experimentData annotation protocolData
## assays(1): exprs
## rownames(198): DIRAS3 MAPK14 ... SQSTM1 KCNJ13
## rowData names(0):
## colnames(79): TCGA-OR-A5J1-01A-11R-A29S-07 TCGA-OR-A5J2-01A-11R-A29S-07
## ... TCGA-PK-A5HA-01A-11R-A29S-07 TCGA-PK-A5HB-01A-11R-A29S-07
## colData names(0):
## equivalently
miniACC[["RNASeq2GeneNorm"]]## class: SummarizedExperiment
## dim: 198 79
## metadata(3): experimentData annotation protocolData
## assays(1): exprs
## rownames(198): DIRAS3 MAPK14 ... SQSTM1 KCNJ13
## rowData names(0):
## colnames(79): TCGA-OR-A5J1-01A-11R-A29S-07 TCGA-OR-A5J2-01A-11R-A29S-07
## ... TCGA-PK-A5HA-01A-11R-A29S-07 TCGA-PK-A5HB-01A-11R-A29S-07
## colData names(0):To preserve the colData during the extraction, see
?getWithColData.
Complete cases
complete.cases() shows which patients have complete data
for all assays:
summary(complete.cases(miniACC))## Mode FALSE TRUE
## logical 49 43The above logical vector could be used for patient subsetting. More
simply, intersectColumns() will select complete cases and
rearrange each ExperimentList element so its columns
correspond exactly to rows of colData in the same
order:
accmatched <- intersectColumns(miniACC)Note, the column names of the assays in accmatched are
not the same because of assay-specific identifiers, but they have been
automatically re-arranged to correspond to the same patients. In these
TCGA assays, the first three - delimited positions
correspond to patient, ie the first patient is
TCGA-OR-A5J2:
colnames(accmatched)## CharacterList of length 5
## [["RNASeq2GeneNorm"]] TCGA-OR-A5J2-01A-11R-A29S-07 ...
## [["gistict"]] TCGA-OR-A5J2-01A-11D-A29H-01 ... TCGA-PK-A5HA-01A-11D-A29H-01
## [["RPPAArray"]] TCGA-OR-A5J2-01A-21-A39K-20 ... TCGA-PK-A5HA-01A-21-A39K-20
## [["Mutations"]] TCGA-OR-A5J2-01A-11D-A29I-10 ... TCGA-PK-A5HA-01A-11D-A29I-10
## [["miRNASeqGene"]] TCGA-OR-A5J2-01A-11R-A29W-13 ...Row names that are common across assays
intersectRows() keeps only rows that are common to each
assay, and aligns them in identical order. For example, to keep only
genes where data are available for RNA-seq, GISTIC copy number, and
somatic mutations:
accmatched2 <- intersectRows(miniACC[, ,
c("RNASeq2GeneNorm", "gistict", "Mutations")])## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 126 sampleMap rows not in names(experiments)
rownames(accmatched2)## CharacterList of length 3
## [["RNASeq2GeneNorm"]] DIRAS3 G6PD KDR ERBB3 AKT1S1 ... RET CDKN2A MACC1 CHGA
## [["gistict"]] DIRAS3 G6PD KDR ERBB3 AKT1S1 ... PREX1 RET CDKN2A MACC1 CHGA
## [["Mutations"]] DIRAS3 G6PD KDR ERBB3 AKT1S1 ... PREX1 RET CDKN2A MACC1 CHGAassay and assays
The assay and assays methods follow
SummarizedExperiment convention. The assay
(singular) method will extract the first element of the
ExperimentList and will return a matrix.
## [1] "matrix" "array"The assays (plural) method will return a
SimpleList of the data with each element being a
matrix.
assays(miniACC)## List of length 5
## names(5): RNASeq2GeneNorm gistict RPPAArray Mutations miRNASeqGeneKey point:
Summary of slots and accessors
Slot in the MultiAssayExperiment can be accessed or set
using the respective accessor functions:
| Slot | Accessor |
|---|---|
ExperimentList |
experiments() |
colData |
colData() and $ * |
sampleMap |
sampleMap() |
metadata |
metadata() |
__*__ The $ operator on a
MultiAssayExperiment returns a single column of the
colData.
Transformation / reshaping
The longForm or wideFormat functions will
“reshape” and combine experiments with each other and with
colData into one DataFrame. These functions
provide compatibility with most of the common R/Bioconductor functions
for regression, machine learning, and visualization.
Note. It is best to remove technical replicates before
performing these operations to minimize the number of NA
values, esp. to wideFormat.
longForm
In long format a single column provides all assay results,
with additional optional colData columns whose values are
repeated as necessary. Here assay is the name of the
ExperimentList element, primary is the patient identifier
(rowname of colData), rowname is the assay rowname (in this
case genes), colname is the assay-specific identifier (column
name), value is the numeric measurement (gene expression, copy
number, presence of a non-silent mutation, etc), and following these are
the vital_status and days_to_death colData columns
that have been added:
## harmonizing input:
## removing 126 sampleMap rows not in names(experiments)## DataFrame with 518 rows and 7 columns
## assay primary rowname colname
## <character> <character> <character> <factor>
## 1 RNASeq2GeneNorm TCGA-OR-A5J1 TP53 TCGA-OR-A5J1-01A-11R-A29S-07
## 2 RNASeq2GeneNorm TCGA-OR-A5J1 CTNNB1 TCGA-OR-A5J1-01A-11R-A29S-07
## 3 RNASeq2GeneNorm TCGA-OR-A5J2 TP53 TCGA-OR-A5J2-01A-11R-A29S-07
## 4 RNASeq2GeneNorm TCGA-OR-A5J2 CTNNB1 TCGA-OR-A5J2-01A-11R-A29S-07
## 5 RNASeq2GeneNorm TCGA-OR-A5J3 TP53 TCGA-OR-A5J3-01A-11R-A29S-07
## ... ... ... ... ...
## 514 Mutations TCGA-PK-A5HA CTNNB1 TCGA-PK-A5HA-01A-11D-A29I-10
## 515 Mutations TCGA-PK-A5HB TP53 TCGA-PK-A5HB-01A-11D-A29I-10
## 516 Mutations TCGA-PK-A5HB CTNNB1 TCGA-PK-A5HB-01A-11D-A29I-10
## 517 Mutations TCGA-PK-A5HC TP53 TCGA-PK-A5HC-01A-11D-A30A-10
## 518 Mutations TCGA-PK-A5HC CTNNB1 TCGA-PK-A5HC-01A-11D-A30A-10
## value vital_status days_to_death
## <numeric> <integer> <integer>
## 1 563.401 1 1355
## 2 5634.467 1 1355
## 3 165.481 1 1677
## 4 62658.391 1 1677
## 5 956.303 0 NA
## ... ... ... ...
## 514 0 0 NA
## 515 0 0 NA
## 516 0 0 NA
## 517 0 0 NA
## 518 0 0 NA
wideFormat
In wide format, each feature from each assay goes in a separate column, with one row per primary identifier (patient). Here, each variable becomes a new column:
wideFormat(miniACC[c("TP53", "CTNNB1"), , ],
colDataCols = c("vital_status", "days_to_death"))## harmonizing input:
## removing 126 sampleMap rows not in names(experiments)## DataFrame with 92 rows and 9 columns
## primary vital_status days_to_death gistict_TP53 gistict_CTNNB1
## <character> <integer> <integer> <numeric> <numeric>
## 1 TCGA-OR-A5J1 1 1355 0 0
## 2 TCGA-OR-A5J2 1 1677 0 1
## 3 TCGA-OR-A5J3 0 NA 0 0
## 4 TCGA-OR-A5J4 1 423 1 0
## 5 TCGA-OR-A5J5 1 365 0 0
## ... ... ... ... ... ...
## 88 TCGA-PK-A5H9 0 NA 0 0
## 89 TCGA-PK-A5HA 0 NA -1 0
## 90 TCGA-PK-A5HB 0 NA -1 -1
## 91 TCGA-PK-A5HC 0 NA 1 1
## 92 TCGA-P6-A5OG 1 383 -1 1
## Mutations_TP53 Mutations_CTNNB1 RNASeq2GeneNorm_TP53 RNASeq2GeneNorm_CTNNB1
## <numeric> <numeric> <numeric> <numeric>
## 1 0 0 563.401 5634.47
## 2 1 1 165.481 62658.39
## 3 0 0 956.303 6337.43
## 4 0 0 NA NA
## 5 0 0 1169.636 5979.06
## ... ... ... ... ...
## 88 0 0 890.866 5258.99
## 89 0 0 683.572 8120.17
## 90 0 0 237.370 5257.81
## 91 0 0 NA NA
## 92 NA NA 815.345 6390.10Key points
- Knowing how to subset a
MultiAssayExperimentobject is important to be able to restrict observations and measurements to particular phenotypes or sample types - Functions such as
longFormandwideFormatare helpful for downstream analysis functions that require a certain type of input format
SingleCellMultiModal
SingleCellMultiModal is an ExperimentHub
package that serves multiple datasets obtained from GEO and other
sources and represents them as MultiAssayExperiment
objects. We provide several multi-modal datasets including scNMT, 10X
Multiome, seqFISH, CITEseq, SCoPE2, and others. The scope of the package
is is to provide data for benchmarking and analysis.
Users can access the data for a particular technology with the
appropriate function. For example, to obtain a small data.frame of what
data is available for scNMT, the user would enter:
scNMT("mouse_gastrulation", dry.run = TRUE, version = '2.0.0')## ah_id mode file_size rdataclass rdatadateadded rdatadateremoved
## 1 EH3753 acc_cgi 21.1 Mb matrix 2020-09-03 <NA>
## 2 EH3754 acc_CTCF 1.2 Mb matrix 2020-09-03 <NA>
## 3 EH3755 acc_DHS 16.2 Mb matrix 2020-09-03 <NA>
## 4 EH3756 acc_genebody 60.1 Mb matrix 2020-09-03 <NA>
## 5 EH3757 acc_p300 0.2 Mb matrix 2020-09-03 <NA>
## 6 EH3758 acc_promoter 33.8 Mb matrix 2020-09-03 <NA>
## 7 EH3760 met_cgi 12.1 Mb matrix 2020-09-03 <NA>
## 8 EH3761 met_CTCF 0.1 Mb matrix 2020-09-03 <NA>
## 9 EH3762 met_DHS 3.9 Mb matrix 2020-09-03 <NA>
## 10 EH3763 met_genebody 33.9 Mb matrix 2020-09-03 <NA>
## 11 EH3764 met_p300 0.1 Mb matrix 2020-09-03 <NA>
## 12 EH3765 met_promoter 18.7 Mb matrix 2020-09-03 <NA>
## 13 EH3766 rna 43.5 Mb matrix 2020-09-03 <NA>We can see that there are a few assays for each modality.
See the SingleCellMultiModal tutorial above for more
information.