SingleCellMultiModal Introduction
2 July 2025
Source:vignettes/SingleCellMultiModal.Rmd
SingleCellMultiModal.RmdInstallation
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("SingleCellMultiModal")Load packages
library(SingleCellMultiModal)
library(MultiAssayExperiment)Introduction
This package introduces a suite of single-cell multimodal landmark
datasets for benchmarking and testing multimodal analysis methods via
the ExperimentHub Bioconductor package. The scope of this
package is to provide efficient access to a selection of curated,
pre-integrated, publicly available landmark datasets for methods
development and benchmarking.
Representation
Users can obtain integrative representations of multiple modalities
as a MultiAssayExperiment, a common core Bioconductor data
structure relied on by dozens of multimodal data analysis packages.
MultiAssayExperiment harmonizes data management of multiple
experimental assays performed on an overlapping set of specimens.
Although originally developed for patient data from multi-omics cancer
studies, the MultiAssayExperiment framework naturally
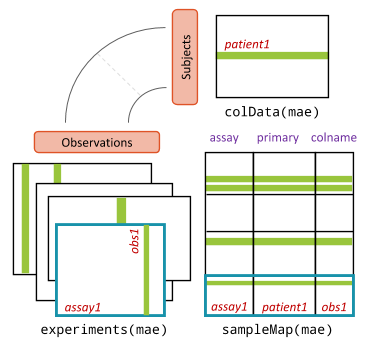
applies also to single cells. A schematic of the data structure can be
seen below. In this context, “patients” are replaced by “cells”. We use
MultiAssayExperiment because it provides a familiar user
experience by extending SummarizedExperiment concepts and
providing open ended compatibility with standard data classes present in
Bioconductor such as the SingleCellExperiment.
Datasets
Here we show a table of available datasets from the
SingleCellMultiModal experiment data package:
DT::datatable(
SingleCellMultiModal::ontomap(),
caption = "Available datasets in SingleCellMultiModal"
)Note that each dataset has its own dedicated function that can also
be invoked with SingleCellMultiModal(). For example, the
SingleCellMultiModal() function can be used to access the
MultiAssayExperiment object for the
mouse_gastrulation dataset:
SingleCellMultiModal::SingleCellMultiModal("mouse_gastrulation")but can also be called individually with the dedicated function,
scNMT() (as seen in the function_name
column).
scNMT(
DataType = "mouse_gastrulation",
modes = "*",
version = "2.0.0",
dry.run = TRUE
) |>
knitr::kable()| ah_id | mode | file_size | rdataclass | rdatadateadded | rdatadateremoved |
|---|---|---|---|---|---|
| EH3753 | acc_cgi | 21.1 Mb | matrix | 2020-09-03 | NA |
| EH3754 | acc_CTCF | 1.2 Mb | matrix | 2020-09-03 | NA |
| EH3755 | acc_DHS | 16.2 Mb | matrix | 2020-09-03 | NA |
| EH3756 | acc_genebody | 60.1 Mb | matrix | 2020-09-03 | NA |
| EH3757 | acc_p300 | 0.2 Mb | matrix | 2020-09-03 | NA |
| EH3758 | acc_promoter | 33.8 Mb | matrix | 2020-09-03 | NA |
| EH3760 | met_cgi | 12.1 Mb | matrix | 2020-09-03 | NA |
| EH3761 | met_CTCF | 0.1 Mb | matrix | 2020-09-03 | NA |
| EH3762 | met_DHS | 3.9 Mb | matrix | 2020-09-03 | NA |
| EH3763 | met_genebody | 33.9 Mb | matrix | 2020-09-03 | NA |
| EH3764 | met_p300 | 0.1 Mb | matrix | 2020-09-03 | NA |
| EH3765 | met_promoter | 18.7 Mb | matrix | 2020-09-03 | NA |
| EH3766 | rna | 43.5 Mb | matrix | 2020-09-03 | NA |
Individual vignettes
To see the technology specific vignettes, use the following command to list the vignettes:
help(package = "SingleCellMultiModal")and click on
in the Help pane of RStudio.
Session Information
Click to expand
## R version 4.5.1 (2025-06-13)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.2 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: Etc/UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] SingleCellMultiModal_1.20.0 MultiAssayExperiment_1.34.0
## [3] SummarizedExperiment_1.38.1 Biobase_2.68.0
## [5] GenomicRanges_1.60.0 GenomeInfoDb_1.44.0
## [7] IRanges_2.42.0 S4Vectors_0.46.0
## [9] BiocGenerics_0.54.0 generics_0.1.4
## [11] MatrixGenerics_1.20.0 matrixStats_1.5.0
## [13] BiocStyle_2.36.0
##
## loaded via a namespace (and not attached):
## [1] tidyselect_1.2.1 dplyr_1.1.4
## [3] blob_1.2.4 filelock_1.0.3
## [5] Biostrings_2.76.0 fastmap_1.2.0
## [7] SingleCellExperiment_1.30.1 BiocFileCache_2.16.0
## [9] promises_1.3.3 digest_0.6.37
## [11] mime_0.13 lifecycle_1.0.4
## [13] KEGGREST_1.48.1 RSQLite_2.4.1
## [15] magrittr_2.0.3 compiler_4.5.1
## [17] rlang_1.1.6 sass_0.4.10
## [19] tools_4.5.1 yaml_2.3.10
## [21] knitr_1.50 S4Arrays_1.8.1
## [23] htmlwidgets_1.6.4 bit_4.6.0
## [25] curl_6.4.0 DelayedArray_0.34.1
## [27] abind_1.4-8 withr_3.0.2
## [29] purrr_1.0.4 desc_1.4.3
## [31] grid_4.5.1 ExperimentHub_2.16.0
## [33] xtable_1.8-4 cli_3.6.5
## [35] rmarkdown_2.29 crayon_1.5.3
## [37] ragg_1.4.0 rjson_0.2.23
## [39] httr_1.4.7 BiocBaseUtils_1.10.0
## [41] DBI_1.2.3 cachem_1.1.0
## [43] AnnotationDbi_1.70.0 BiocManager_1.30.26
## [45] XVector_0.48.0 vctrs_0.6.5
## [47] Matrix_1.7-3 jsonlite_2.0.0
## [49] bookdown_0.43 bit64_4.6.0-1
## [51] crosstalk_1.2.1 magick_2.8.7
## [53] systemfonts_1.2.3 jquerylib_0.1.4
## [55] glue_1.8.0 pkgdown_2.1.3
## [57] DT_0.33 BiocVersion_3.21.1
## [59] later_1.4.2 UCSC.utils_1.4.0
## [61] tibble_3.3.0 pillar_1.10.2
## [63] rappdirs_0.3.3 htmltools_0.5.8.1
## [65] GenomeInfoDbData_1.2.14 R6_2.6.1
## [67] dbplyr_2.5.0 textshaping_1.0.1
## [69] evaluate_1.0.4 shiny_1.11.0
## [71] lattice_0.22-7 AnnotationHub_3.16.0
## [73] png_0.1-8 SpatialExperiment_1.18.1
## [75] memoise_2.0.1 httpuv_1.6.16
## [77] bslib_0.9.0 Rcpp_1.0.14
## [79] SparseArray_1.8.0 xfun_0.52
## [81] fs_1.6.6 pkgconfig_2.0.3