Data integration and stats
Source:vignettes/articles/data_integration_and_stats.Rmd
data_integration_and_stats.Rmd
library(bugphyzzAnalyses)
library(bugphyzz)
library(dplyr)
library(taxPPro)
library(ggplot2)
library(forcats)
library(ggpubr)
library(taxPPro)
library(tidyr)
library(ComplexHeatmap)
library(purrr)
library(forcats)
library(ggbreak)
library(tibble)
library(patchwork)
library(grid)
library(cowplot)Bugphyzz data
discrete_types <- c(
"binary", "multistate-intersection", "multistate-union"
)
bp <- importBugphyzz(v = 0.5) |>
map(~ mutate(.x, NCBI_ID = as.character(NCBI_ID))) |>
map(~ {
attr_type <- unique(.x$Attribute_type)
if (attr_type %in% discrete_types) {
df <- .x |>
filter(
!(Validation < 0.7 & Evidence == "asr")
)
} else if (attr_type == "numeric") {
df <- .x |>
filter(
!(Validation < 0.5 & Evidence == "asr")
)
}
df
})
length(bp)
#> [1] 32
myDataTable(data.frame(attributes = names(bp)), page_len = length(bp))Totals
## Number of unique taxa
n_taxa <- map(bp, ~ .x$NCBI_ID) |>
unlist(use.names = FALSE) |>
unique() |>
length()
## Number of annotations
n_annotations <- map_int(bp, nrow) |>
sum()
## Number of physiologies/attribute groups
n_attr <- length(bp)
## Number of sources
n_sources <- map(bp, ~ {
unique(.x$Attribute_source) |>
{\(y) y[!is.na(y)]}()
}) |>
unlist(recursive = TRUE, use.names = FALSE) |>
unique() |>
length()
## Number of annotations from sources
n_annotations_source <- map_int(bp, ~ {
.x |>
filter(!is.na(Attribute_source)) |>
nrow()
}) |>
sum()
## Number of annotations IBD
n_annotations_ibd <- map_int(bp, ~ {
.x |>
filter(Evidence == "tax") |>
nrow()
}) |>
sum()
## Number of annotations ASR
n_annotations_asr <- map_int(bp, ~ {
.x |>
filter(Evidence == "asr") |>
nrow()
}) |>
sum()
## Number of
num_vs_dis_n <- map_chr(bp, ~ {
unique(.x$Attribute_type)
}) |>
table() |>
as.data.frame() |>
set_names(c("name", "n")) |>
mutate(name = ifelse(name == "numeric", "numeric", "discrete")) |>
count(name, wt = n)
attr_asr_default_names <- map(bp, ~ {
unique(.x$Evidence)
}) |>
keep(~ "asr" %in% .x) |>
names()
attr_asr_default <- length(attr_asr_default_names)
attr_asr_all <- bugphyzz:::.validationData() |>
filter(rank == "all") |>
select(physiology, mcc_mean, r2_mean) |>
pull(physiology) |>
unique() |>
length()
count_summary_tbl <- tibble::tribble(
~name, ~n,
"Number of taxa", n_taxa,
"Number of annotations", n_annotations,
"Number of attributes", n_attr,
"Number of discrete attributes", pull(filter(num_vs_dis_n, name == "discrete"),n),
"Number of sources", n_sources,
"Number of annoations from sources", n_annotations_source,
"Number of IBD annotations", n_annotations_ibd,
"Number of annotations imputed through ASR", n_annotations_asr,
"Number of attributes with ASR data (default > 0.5 val)", attr_asr_default,
"Number of attributes with ASR data (all)", attr_asr_all
)
myDataTable(count_summary_tbl)Totals by rank
valid_ranks <- c(
"superkingdom", "phylum", "class", "order", "family",
"genus", "species", "strain"
)
attr_order <- names(sort(map_int(bp, nrow), decreasing = TRUE))
counts_rank <- bp |>
map(~ count(.x, Attribute, Rank)) |>
bind_rows() |>
mutate(Attribute = factor(Attribute, levels = attr_order)) |>
mutate(Rank = factor(Rank, levels = valid_ranks))
p_counts_rank <- counts_rank |>
ggplot(aes(Attribute, n)) +
geom_col(aes(fill = Rank), position = "stack") +
scale_fill_discrete(name = "Rank") +
scale_y_continuous(labels = scales::comma) +
labs(
x = "Attribute", y = "No. annotations"
) +
theme_bw() +
theme(
axis.text.x = element_text(angle = 45, hjust = 1)
)
p_counts_rank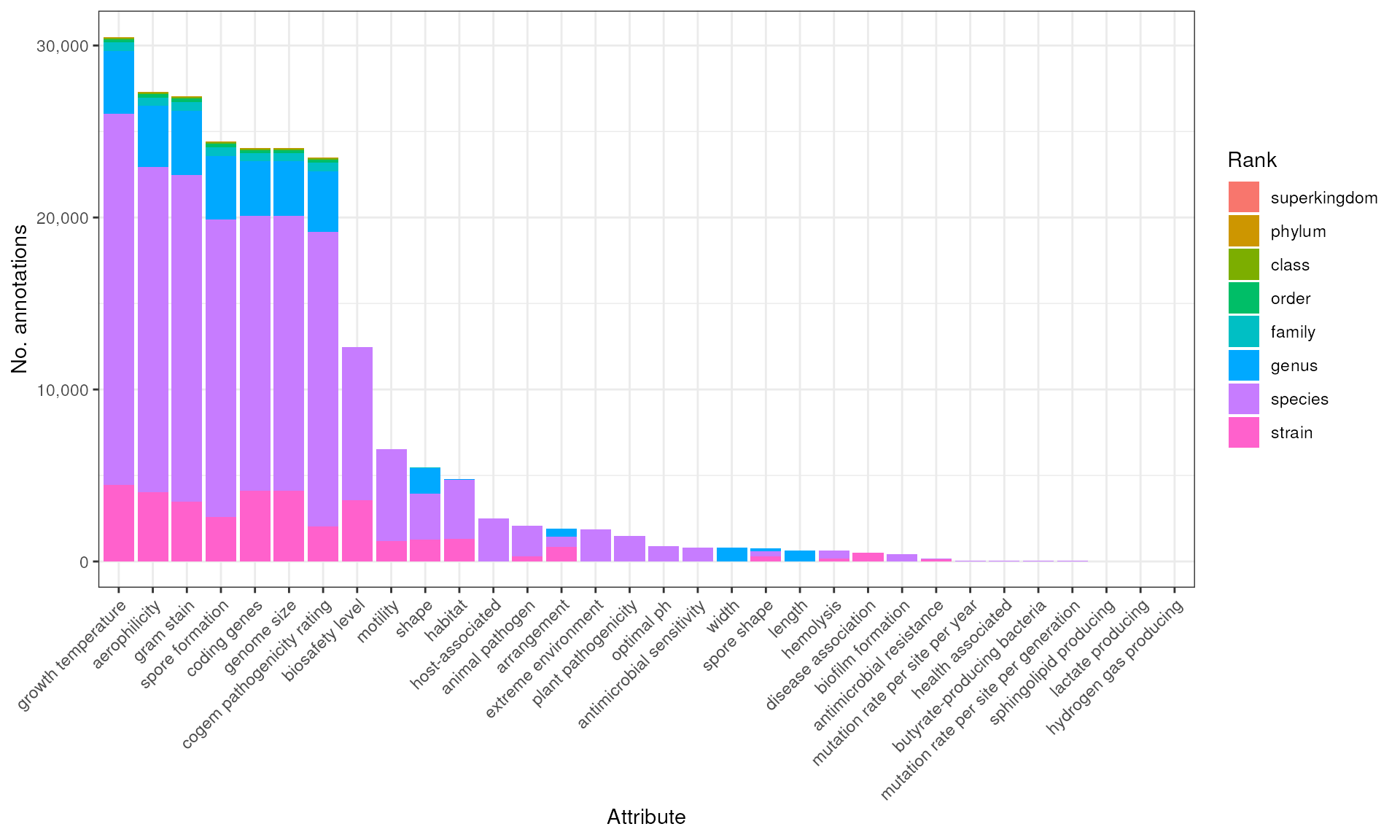
Total number of annotations by type of Evidence
count_types <- bp |>
map (~{
.x |>
mutate(
Type = case_when(
Evidence %in% c("exp", "igc", "nas", "tas") ~ "Source",
Evidence == "asr" ~ "ASR",
Evidence == "tax" ~ "IBD"
)
) |>
count(Attribute, Type)
}) |>
bind_rows()
keep_attrs <- split(count_types, count_types$Attribute) |>
keep( ~ {
types <- unique(.x$Type)
"IBD" %in% types | "ASR" %in% types
}) |>
names()
p_count_types <- count_types |>
filter(Attribute %in% keep_attrs) |>
mutate(Attribute = factor(Attribute, levels = attr_order)) |>
ggplot(aes(Attribute, n)) +
geom_col(aes(fill = Type), position = "stack") +
labs(
x = "Attribute", y = "No. annotations"
) +
scale_fill_discrete(name = "Annotation types") +
scale_y_continuous(labels = scales::comma) +
theme_bw() +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
p_count_types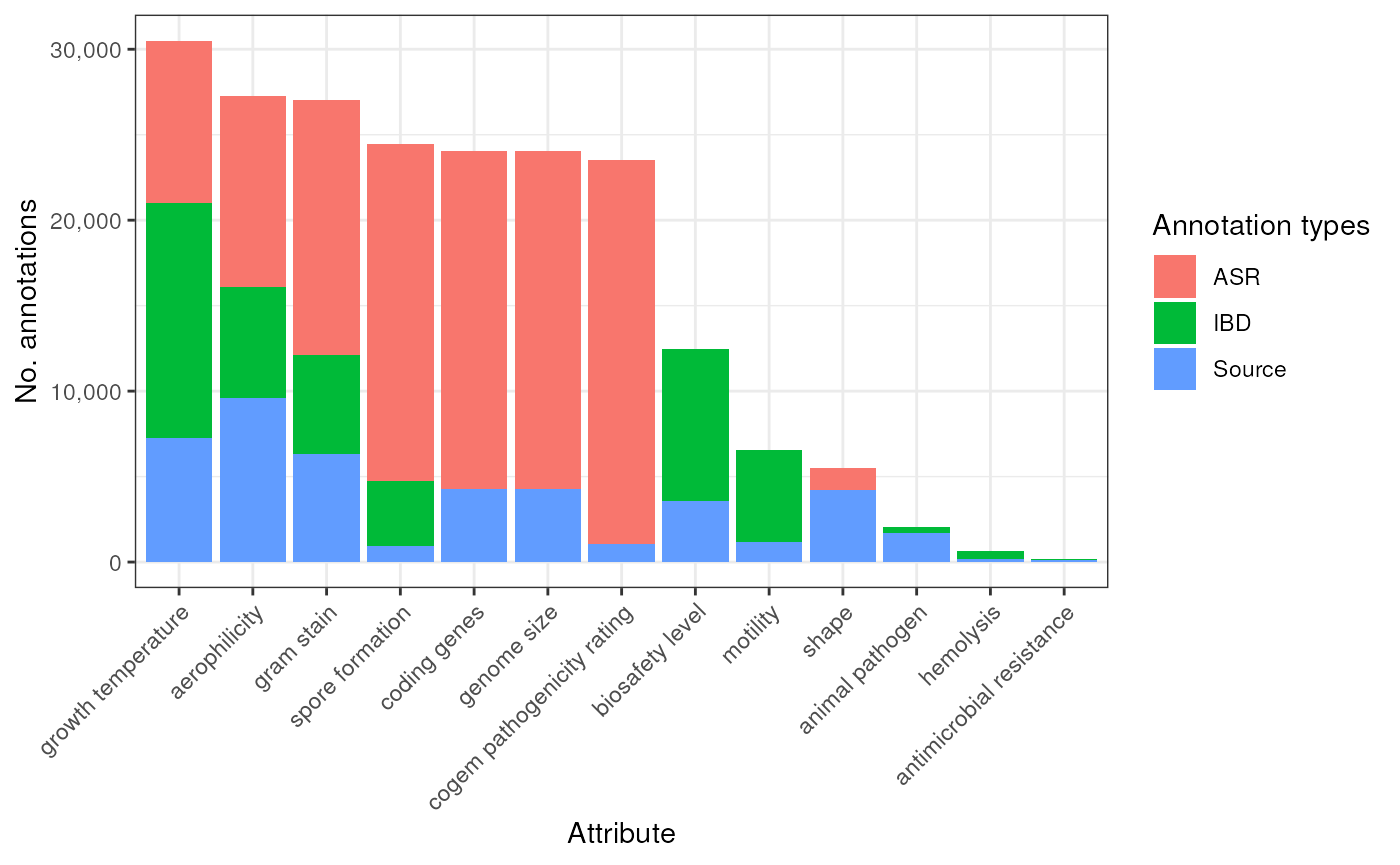
Figure: 10-fold cross-validation and counts per evidence type (species)
Create plot of MMC values for discrete attributes:
# val_fname <- "/home/samuelgamboa/Projects/taxPProValidation/validation_summary.tsv"
# val <- readr::read_tsv(val_fname, show_col_types = FALSE) |>
# filter(rank == "all")
val <- bugphyzz:::.validationData() |>
filter(rank == "all") # Validation including taxa of mixed ranks as input (keyword is "all")
dis_phys_names <- val |>
filter(!is.na(mcc_mean)) |>
group_by(physiology) |>
slice_max(order_by = mcc_mean, n = 1) |>
arrange(-mcc_mean) |>
pull(physiology)
pValDis <- val |> # plot Validation Discrete
filter(!is.na(mcc_mean)) |>
mutate(ltp_bp = ifelse(is.na(ltp_bp), ltp_bp_phys, ltp_bp)) |>
mutate(physiology = factor(physiology, levels = dis_phys_names)) |>
mutate(
guide_col = ifelse(
as.character(physiology) == attribute,
as.character(physiology),
paste0(as.character(physiology), "|", attribute)
)
) |>
arrange(physiology, mcc_mean) |>
ggplot(aes(forcats::fct_inorder(guide_col), mcc_mean, group = attribute)) +
geom_errorbar(
aes(ymin = mcc_mean - mcc_sd, ymax = mcc_mean + mcc_sd),
position = position_dodge(0.5),
width = 0, linewidth = 0.3
) +
geom_hline(
yintercept = 0.7, linetype = "dotdash", linewidth = 0.2,
color = "red"
) +
geom_point(
aes(size = ltp_bp),
shape = 23, position = position_dodge(0.5), fill = "white"
# size = 2
) +
scale_size_continuous(name = "tips annotated") +
scale_y_continuous(
breaks = round(
seq(
min(val$mcc_mean - val$mcc_sd, na.rm = TRUE) - 0.1,
max(val$mcc_mean + val$mcc_sd, na.rm = TRUE) + 0.1,
0.2),
1)
) +
labs(
y = "Matthews Correlation Coefficient",
x = "Attribute|Attribute value"
) +
theme_bw() +
theme(
panel.grid.minor = element_blank()
) +
coord_flip()Create plot of R-squared values for continuous attributes:
num <- val |>
filter(!is.na(r2_mean))
num_phys_names <- num |>
group_by(physiology) |>
slice_max(order_by = r2_mean, n = 1) |>
arrange(-r2_mean) |>
pull(physiology)
pValNum <- val |> # plot Validation Numeric attributes
filter(!is.na(r2_mean)) |>
mutate(ltp_bp = ifelse(is.na(ltp_bp), ltp_bp_phys, ltp_bp)) |>
ggplot(aes(reorder(physiology, -r2_mean), r2_mean, group = attribute)) +
geom_errorbar(
aes(ymin = r2_mean - r2_sd, ymax = r2_mean + r2_sd),
position = position_dodge(0.5),
width = 0, linewidth = 0.3
) +
geom_hline(
yintercept = 0.5, linetype = "dotdash", linewidth = 0.2,
color = "red"
) +
geom_point(
aes(size = ltp_bp),
shape = 23, position = position_dodge(0.5), fill = "white"
# size = 2
) +
scale_size_continuous(name = "tips annotated") +
scale_y_continuous(
breaks = round(seq(min(val$r2_mean - val$r2_sd, na.rm = TRUE) - 0.1, max(val$r2_mean + val$r2_sd, na.rm = TRUE) + 0.1, 0.2), 1)
) +
labs(
y = "R-squared",
x = "Attribute"
) +
theme_bw() +
theme(
panel.grid.minor = element_blank()
) +
coord_flip()Crete plot of counts of annotations at the species level. Stack colors/columns with colors according to Evidence type of the annotations: ASR, IBD, Source. Only include attributes that were expanded with IBD or ASR.
counts_type_sp <- bp |>
map(~ {
.x |>
filter(Rank == "species") |>
mutate(
Evidence = case_when(
Evidence == "tax" ~ "IBD",
Evidence == "asr" ~ "ASR",
Evidence %in% c("exp", "igc", "tas", "nas") ~ "Source",
TRUE ~ Evidence
)
) |>
count(Attribute, Evidence) |>
mutate(total = sum(n))
}) |>
bind_rows()
p_counts_sp <- counts_type_sp |>
filter(Attribute %in% keep_attrs) |>
mutate(Evidence = factor(Evidence, levels = c("ASR", "IBD", "Source"))) |>
ggplot(aes(reorder(Attribute, total), n)) +
geom_col(aes(fill = Evidence), position = "stack") +
labs(
y = "No. annotations (species)", x = "Attribute"
) +
scale_fill_manual(
values = c("#56B4E9", "#D55E00", "#0072B2"),
name = "Annotation type"
) +
scale_y_continuous(labels = scales::comma) +
theme_bw() +
coord_flip()Merge the plots from the validation results and the counts of annotations
myPlot2 <- plot_grid(
pValNum, p_counts_sp, ncol = 1,
labels = c("b)", "c)"),
rel_heights = c(0.2, 0.8), align = "v"
)
myPlot1 <- plot_grid(
pValDis, myPlot2,
labels = c("a)"),
rel_widths = c(0.55, 0.45), axis = "b"
)
myPlot1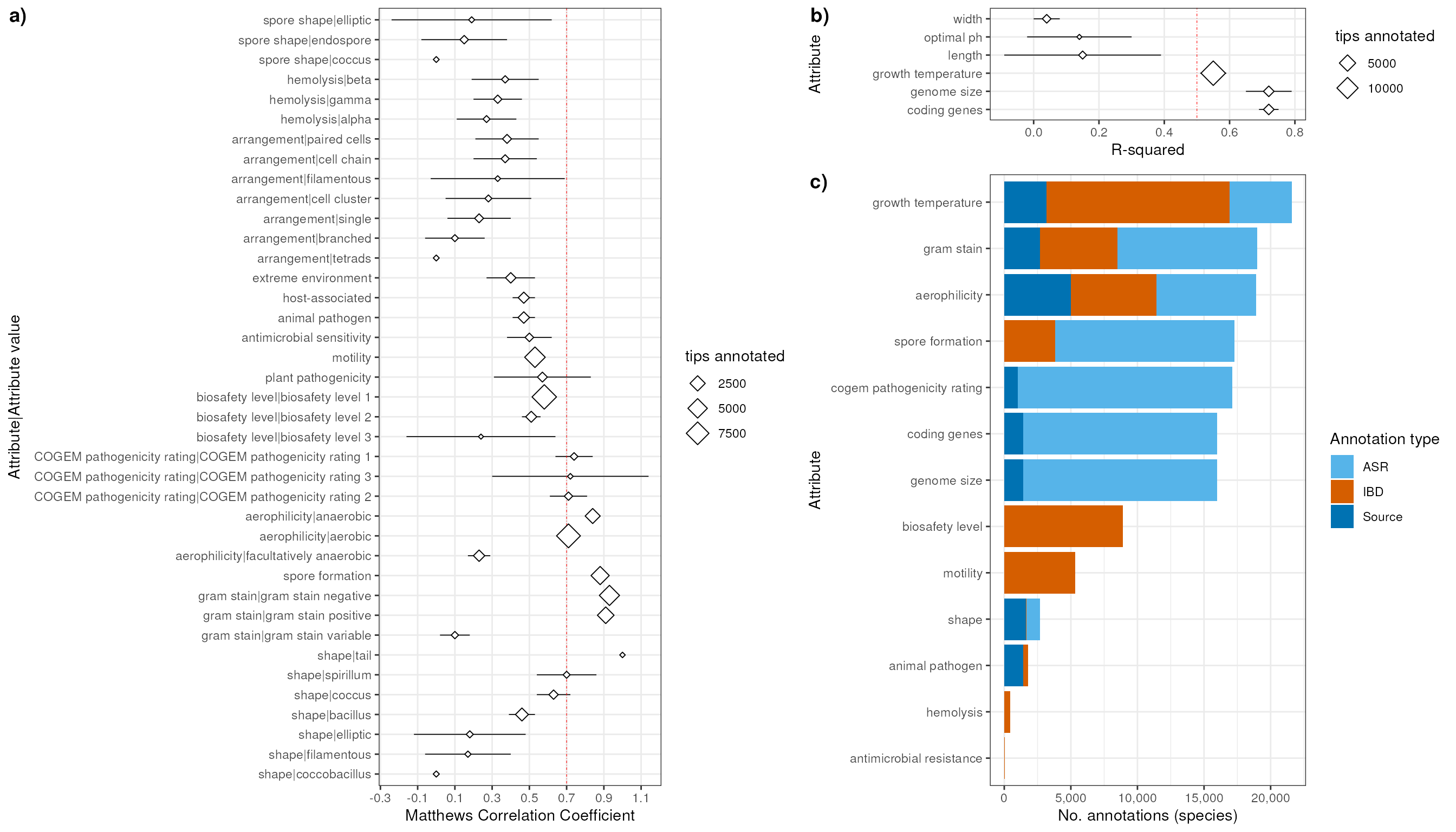
myDataTable(counts_type_sp)Supplementary Figure: Counts of annotations for genus, spescies, and strain and number of annotations per taxon
Number of annotations at the genus, species, and strain level:
counts_type_2 <- bp |>
map(~ {
.x |>
filter(Rank %in% c("genus", "species", "strain")) |>
mutate(
Evidence = case_when(
Evidence == "tax" ~ "IBD",
Evidence == "asr" ~ "ASR",
Evidence %in% c("exp", "igc", "tas", "nas") ~ "Source",
TRUE ~ Evidence
)
) |>
count(Attribute, Rank, Evidence) |>
mutate(total = sum(n))
}) |>
bind_rows()
p_counts_2 <- counts_type_2 |>
filter(Attribute %in% keep_attrs) |>
mutate(Evidence = factor(Evidence, levels = c("ASR", "IBD", "Source"))) |>
ggplot(aes(reorder(Attribute, total), n)) +
geom_col(aes(fill = Evidence), position = "stack") +
labs(
y = "No. annotations", x = "Attribute"
) +
scale_fill_manual(
values = c("#56B4E9", "#D55E00", "#0072B2"),
name = "Annotation type"
) +
scale_y_continuous(labels = scales::comma) +
theme_bw() +
facet_wrap(~Rank, nrow = 1, scales = "free_x") +
theme(
axis.text.x = element_text(angle = 45, hjust = 1)
) +
coord_flip()Frequency of number of annotations per single taxon:
n_annot_tax <- bp |>
map(~ {
attr_type <- unique(.x$Attribute_type)
.x |>
filter(Rank %in% c("strain", "species", "genus")) |>
select(Attribute, NCBI_ID, Rank)
}) |>
bind_rows() |>
count(NCBI_ID, Rank)
p_n_annot_tax <- n_annot_tax |>
ggplot() +
geom_histogram(aes(x = n), binwidth = 1) +
labs(
x = "Annotations per\ntaxon",
y = "Frequency"
) +
facet_wrap(~Rank, ncol = 1) +
theme_bw()Merge plots above for supplementary figure:
suppN <- plot_grid(
p_counts_2, p_n_annot_tax,
labels = c("a)", "b)"),
rel_widths = c(0.8, 0.20),
nrow = 1
)
suppN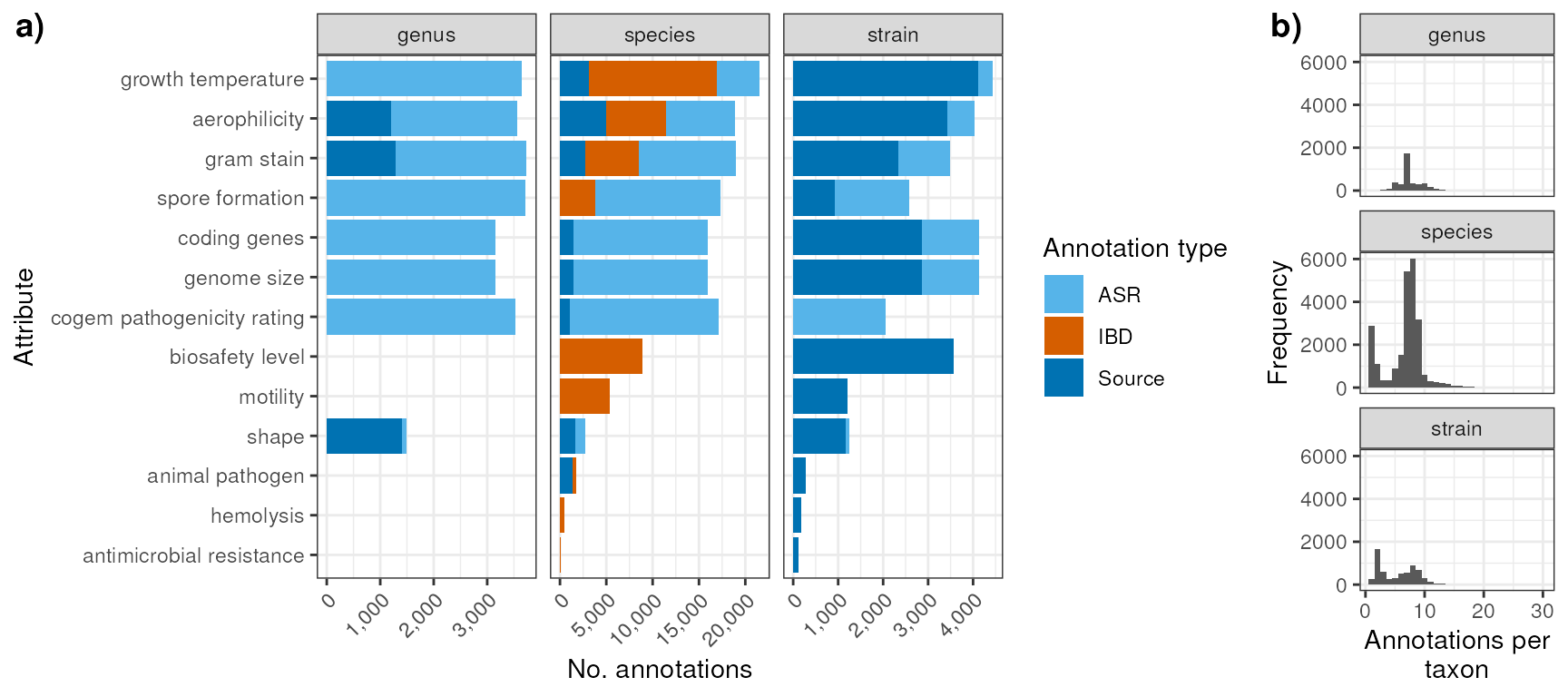
Figure: Heatmap Sources x Attribute x Rank
mySources <- bp |>
map(~ {
.x |>
select(Attribute_source, Confidence_in_curation, Evidence) |>
drop_na() |>
rename(Confidence = Confidence_in_curation)
}) |>
bind_rows() |>
distinct() |>
mutate(
Confidence_color = case_when(
Confidence == "high" ~ "red",
Confidence == "medium" ~ "orange",
Confidence == "low" ~ "yellow"
),
Evindence_color = case_when(
Evidence == "exp" ~ "blue",
Evidence == "igc" ~ "green",
Evidence == "tas" ~ "brown4",
Evidence == "nas" ~ "purple"
),
Confidence = factor(Confidence, levels = c("high", "medium", "low")),
Evidence = factor(Evidence, levels = c("exp", "igc", "tas", "nas"))
)
source_attribute_rank <- bp |>
map(~{
.x |>
filter(!is.na(Attribute_source)) |>
count(Attribute, Attribute_source, Rank)
}) |>
bind_rows()
mat <- source_attribute_rank |>
arrange(Attribute, Rank) |>
unite(col = "Attribute|Rank", Attribute, Rank, sep = "|") |>
pivot_wider(
names_from = "Attribute_source", values_from = "n"
) |>
tibble::column_to_rownames(var = "Attribute|Rank") |>
as.data.frame() |>
as.matrix()
mat[is.na(mat)] <- 0
mat[mat > 0] <- 1
color_fun <- circlize::colorRamp2(
breaks = c(0, 1), colors = c('white', "gray20")
)
rankAnnotationsDF <- data.frame(
Rank = sub("^(.*\\|)(\\w+)$", "\\2", rownames(mat))
)
rank_colors =list(
Rank = c("genus" = "orange","species" = "#009E73", "strain" = "#CC79A7")
)
row_ha <- rowAnnotation(
df = rankAnnotationsDF, col = rank_colors, width = unit(5, "cm")
)
mySources <- mySources[match(colnames(mat), mySources$Attribute_source),]
myColColors <- list(
Confidence = c(high = "red", medium = "orange", low = "yellow"),
Evidence = c(exp = "blue", igc = "green", tas = "pink3", nas = "purple")
)
column_ha <- columnAnnotation(
df = mySources[, c("Confidence", "Evidence")],
col = myColColors,
height = unit(5, "cm")
)
splits_attrs <- sub("^(.*)\\|\\w+$", "\\1", rownames(mat))
hp <- Heatmap(
matrix = mat,
cluster_rows = FALSE,
cluster_columns = FALSE,
show_row_names = FALSE,
show_heatmap_legend = TRUE,
col = color_fun(c(0, 1)),
name = "Annotation presence",
left_annotation = row_ha,
bottom_annotation = column_ha,
row_split = splits_attrs,
row_title_rot = 0,
heatmap_legend_param = list(
color_bar = "discrete",
border = "black"
),
border = TRUE,
rect_gp = gpar(col = "gray50", lwd = 1),
column_names_max_height = unit(7, "cm")
)
hp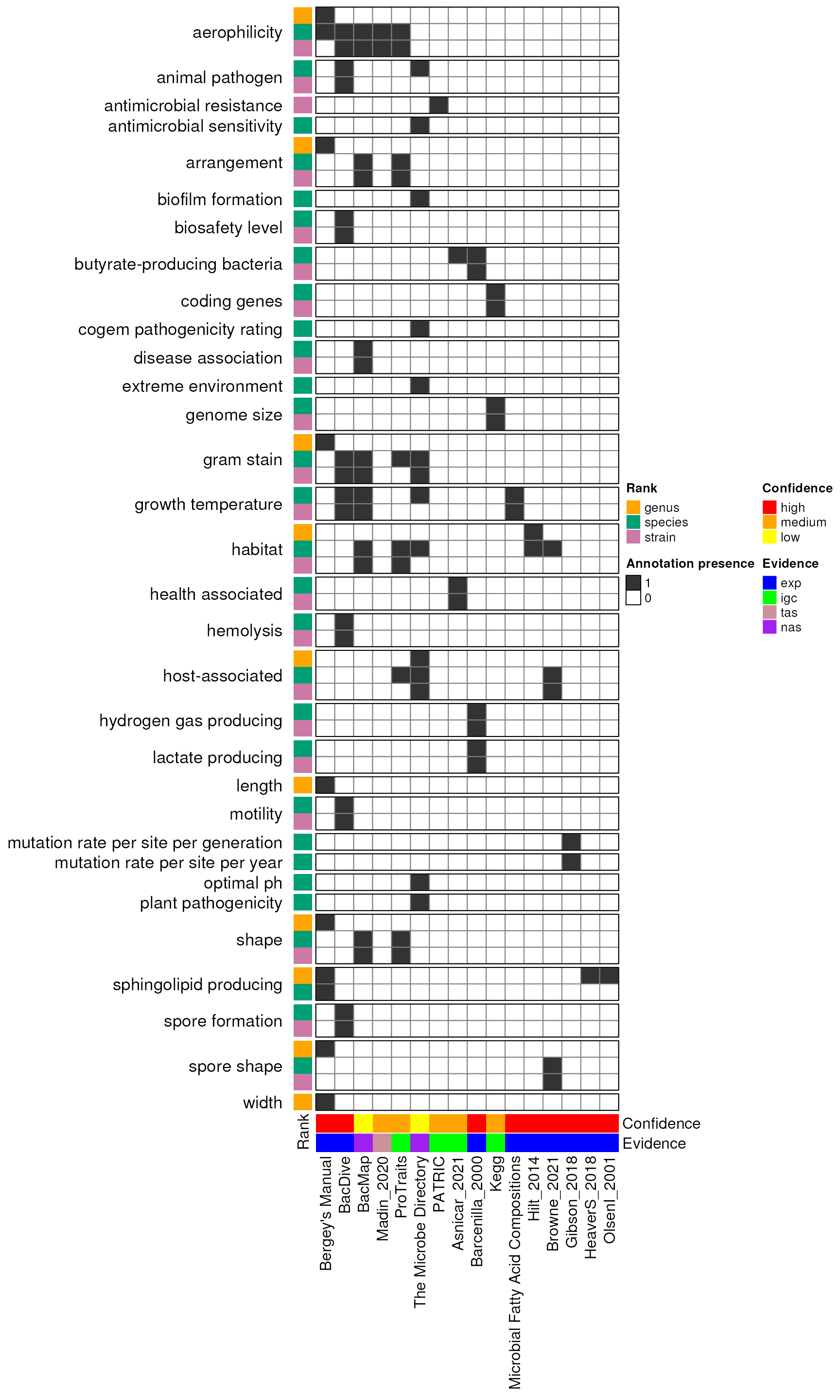
Table/Sup Table: Sources
sources_fname <- system.file(
"extdata", "attribute_sources.tsv", package = "bugphyzz", mustWork = TRUE
)
sources <- read.table(
file = sources_fname,
header = TRUE, sep = "\t", quote = ""
) |>
rename(
`Source (short)` = Attribute_source,
`Confidence in curation` = Confidence_in_curation,
`Source (long)` = full_source
)
myDataTable(sources, page_len = nrow(sources))Session information
sessioninfo::session_info()
#> ─ Session info ───────────────────────────────────────────────────────────────
#> setting value
#> version R version 4.4.1 (2024-06-14)
#> os Ubuntu 22.04.4 LTS
#> system x86_64, linux-gnu
#> ui X11
#> language en
#> collate en_US.UTF-8
#> ctype en_US.UTF-8
#> tz Etc/UTC
#> date 2024-06-28
#> pandoc 3.2 @ /usr/bin/ (via rmarkdown)
#>
#> ─ Packages ───────────────────────────────────────────────────────────────────
#> package * version date (UTC) lib source
#> abind 1.4-5 2016-07-21 [1] RSPM (R 4.4.0)
#> aplot 0.2.3 2024-06-17 [1] RSPM (R 4.4.0)
#> backports 1.5.0 2024-05-23 [1] RSPM (R 4.4.0)
#> Biobase * 2.64.0 2024-04-30 [1] Bioconductor 3.19 (R 4.4.1)
#> BiocFileCache 2.12.0 2024-04-30 [1] Bioconductor 3.19 (R 4.4.1)
#> BiocGenerics * 0.50.0 2024-04-30 [1] Bioconductor 3.19 (R 4.4.1)
#> bit 4.0.5 2022-11-15 [1] RSPM (R 4.4.0)
#> bit64 4.0.5 2020-08-30 [1] RSPM (R 4.4.0)
#> blob 1.2.4 2023-03-17 [1] RSPM (R 4.4.0)
#> broom 1.0.6 2024-05-17 [1] RSPM (R 4.4.0)
#> bslib 0.7.0 2024-03-29 [1] RSPM (R 4.4.0)
#> bugphyzz * 0.99.3 2024-06-28 [1] Github (waldronlab/bugphyzz@8fc1c6d)
#> bugphyzzAnalyses * 0.1.19 2024-06-28 [1] local
#> cachem 1.1.0 2024-05-16 [1] RSPM (R 4.4.0)
#> Cairo 1.6-2 2023-11-28 [1] RSPM (R 4.4.0)
#> car 3.1-2 2023-03-30 [1] RSPM (R 4.4.0)
#> carData 3.0-5 2022-01-06 [1] RSPM (R 4.4.0)
#> circlize 0.4.16 2024-02-20 [1] RSPM (R 4.4.0)
#> cli 3.6.3 2024-06-21 [1] RSPM (R 4.4.0)
#> clue 0.3-65 2023-09-23 [1] RSPM (R 4.4.0)
#> cluster 2.1.6 2023-12-01 [2] CRAN (R 4.4.1)
#> codetools 0.2-20 2024-03-31 [2] CRAN (R 4.4.1)
#> colorspace 2.1-0 2023-01-23 [1] RSPM (R 4.4.0)
#> ComplexHeatmap * 2.20.0 2024-04-30 [1] Bioconductor 3.19 (R 4.4.1)
#> cowplot * 1.1.3 2024-01-22 [1] RSPM (R 4.4.0)
#> crayon 1.5.3 2024-06-20 [1] RSPM (R 4.4.0)
#> crosstalk 1.2.1 2023-11-23 [1] RSPM (R 4.4.0)
#> curl 5.2.1 2024-03-01 [1] RSPM (R 4.4.0)
#> DBI 1.2.3 2024-06-02 [1] RSPM (R 4.4.0)
#> dbplyr 2.5.0 2024-03-19 [1] RSPM (R 4.4.0)
#> DelayedArray 0.30.1 2024-05-07 [1] Bioconductor 3.19 (R 4.4.1)
#> desc 1.4.3 2023-12-10 [1] RSPM (R 4.4.0)
#> digest 0.6.36 2024-06-23 [1] RSPM (R 4.4.0)
#> doParallel 1.0.17 2022-02-07 [1] RSPM (R 4.4.0)
#> dplyr * 1.1.4 2023-11-17 [1] RSPM (R 4.4.0)
#> DT 0.33 2024-04-04 [1] RSPM (R 4.4.0)
#> evaluate 0.24.0 2024-06-10 [1] RSPM (R 4.4.0)
#> fansi 1.0.6 2023-12-08 [1] RSPM (R 4.4.0)
#> farver 2.1.2 2024-05-13 [1] RSPM (R 4.4.0)
#> fastmap 1.2.0 2024-05-15 [1] RSPM (R 4.4.0)
#> filelock 1.0.3 2023-12-11 [1] RSPM (R 4.4.0)
#> forcats * 1.0.0 2023-01-29 [1] RSPM (R 4.4.0)
#> foreach 1.5.2 2022-02-02 [1] RSPM (R 4.4.0)
#> fs 1.6.4 2024-04-25 [1] RSPM (R 4.4.0)
#> generics 0.1.3 2022-07-05 [1] RSPM (R 4.4.0)
#> GenomeInfoDb * 1.40.1 2024-05-24 [1] Bioconductor 3.19 (R 4.4.1)
#> GenomeInfoDbData 1.2.12 2024-06-25 [1] Bioconductor
#> GenomicRanges * 1.56.1 2024-06-12 [1] Bioconductor 3.19 (R 4.4.1)
#> GetoptLong 1.0.5 2020-12-15 [1] RSPM (R 4.4.0)
#> ggbreak * 0.1.2 2023-06-26 [1] RSPM (R 4.4.0)
#> ggfun 0.1.5 2024-05-28 [1] RSPM (R 4.4.0)
#> ggplot2 * 3.5.1 2024-04-23 [1] RSPM (R 4.4.0)
#> ggplotify 0.1.2 2023-08-09 [1] RSPM (R 4.4.0)
#> ggpubr * 0.6.0 2023-02-10 [1] RSPM (R 4.4.0)
#> ggsignif 0.6.4 2022-10-13 [1] RSPM (R 4.4.0)
#> GlobalOptions 0.1.2 2020-06-10 [1] RSPM (R 4.4.0)
#> glue 1.7.0 2024-01-09 [1] RSPM (R 4.4.0)
#> gridGraphics 0.5-1 2020-12-13 [1] RSPM (R 4.4.0)
#> gtable 0.3.5 2024-04-22 [1] RSPM (R 4.4.0)
#> highr 0.11 2024-05-26 [1] RSPM (R 4.4.0)
#> htmltools 0.5.8.1 2024-04-04 [1] RSPM (R 4.4.0)
#> htmlwidgets 1.6.4 2023-12-06 [1] RSPM (R 4.4.0)
#> httr 1.4.7 2023-08-15 [1] RSPM (R 4.4.0)
#> httr2 1.0.1 2024-04-01 [1] RSPM (R 4.4.0)
#> IRanges * 2.38.0 2024-04-30 [1] Bioconductor 3.19 (R 4.4.1)
#> iterators 1.0.14 2022-02-05 [1] RSPM (R 4.4.0)
#> jquerylib 0.1.4 2021-04-26 [1] RSPM (R 4.4.0)
#> jsonlite 1.8.8 2023-12-04 [1] RSPM (R 4.4.0)
#> knitr 1.47 2024-05-29 [1] RSPM (R 4.4.0)
#> labeling 0.4.3 2023-08-29 [1] RSPM (R 4.4.0)
#> lattice 0.22-6 2024-03-20 [2] CRAN (R 4.4.1)
#> lifecycle 1.0.4 2023-11-07 [1] RSPM (R 4.4.0)
#> magick 2.8.3 2024-02-18 [1] RSPM (R 4.4.0)
#> magrittr 2.0.3 2022-03-30 [1] RSPM (R 4.4.0)
#> Matrix 1.7-0 2024-04-26 [2] CRAN (R 4.4.1)
#> MatrixGenerics * 1.16.0 2024-04-30 [1] Bioconductor 3.19 (R 4.4.1)
#> matrixStats * 1.3.0 2024-04-11 [1] RSPM (R 4.4.0)
#> memoise 2.0.1 2021-11-26 [1] RSPM (R 4.4.0)
#> munsell 0.5.1 2024-04-01 [1] RSPM (R 4.4.0)
#> patchwork * 1.2.0 2024-01-08 [1] RSPM (R 4.4.0)
#> pillar 1.9.0 2023-03-22 [1] RSPM (R 4.4.0)
#> pkgconfig 2.0.3 2019-09-22 [1] RSPM (R 4.4.0)
#> pkgdown 2.0.9 2024-04-18 [1] RSPM (R 4.4.0)
#> png 0.1-8 2022-11-29 [1] RSPM (R 4.4.0)
#> purrr * 1.0.2 2023-08-10 [1] RSPM (R 4.4.0)
#> R6 2.5.1 2021-08-19 [1] RSPM (R 4.4.0)
#> ragg 1.3.2 2024-05-15 [1] RSPM (R 4.4.0)
#> rappdirs 0.3.3 2021-01-31 [1] RSPM (R 4.4.0)
#> RColorBrewer 1.1-3 2022-04-03 [1] RSPM (R 4.4.0)
#> Rcpp 1.0.12 2024-01-09 [1] RSPM (R 4.4.0)
#> rjson 0.2.21 2022-01-09 [1] RSPM (R 4.4.0)
#> rlang 1.1.4 2024-06-04 [1] RSPM (R 4.4.0)
#> rmarkdown 2.27 2024-05-17 [1] RSPM (R 4.4.0)
#> RSQLite 2.3.7 2024-05-27 [1] RSPM (R 4.4.0)
#> rstatix 0.7.2 2023-02-01 [1] RSPM (R 4.4.0)
#> S4Arrays 1.4.1 2024-05-20 [1] Bioconductor 3.19 (R 4.4.1)
#> S4Vectors * 0.42.0 2024-04-30 [1] Bioconductor 3.19 (R 4.4.1)
#> sass 0.4.9 2024-03-15 [1] RSPM (R 4.4.0)
#> scales 1.3.0 2023-11-28 [1] RSPM (R 4.4.0)
#> sessioninfo 1.2.2 2021-12-06 [1] RSPM (R 4.4.0)
#> shape 1.4.6.1 2024-02-23 [1] RSPM (R 4.4.0)
#> SparseArray 1.4.8 2024-05-24 [1] Bioconductor 3.19 (R 4.4.1)
#> stringi 1.8.4 2024-05-06 [1] RSPM (R 4.4.0)
#> stringr 1.5.1 2023-11-14 [1] RSPM (R 4.4.0)
#> SummarizedExperiment * 1.34.0 2024-05-01 [1] Bioconductor 3.19 (R 4.4.1)
#> systemfonts 1.1.0 2024-05-15 [1] RSPM (R 4.4.0)
#> taxPPro * 1.0.0 2024-06-28 [1] Github (waldronlab/taxPPro@b0460fe)
#> textshaping 0.4.0 2024-05-24 [1] RSPM (R 4.4.0)
#> tibble * 3.2.1 2023-03-20 [1] RSPM (R 4.4.0)
#> tidyr * 1.3.1 2024-01-24 [1] RSPM (R 4.4.0)
#> tidyselect 1.2.1 2024-03-11 [1] RSPM (R 4.4.0)
#> UCSC.utils 1.0.0 2024-04-30 [1] Bioconductor 3.19 (R 4.4.1)
#> utf8 1.2.4 2023-10-22 [1] RSPM (R 4.4.0)
#> vctrs 0.6.5 2023-12-01 [1] RSPM (R 4.4.0)
#> withr 3.0.0 2024-01-16 [1] RSPM (R 4.4.0)
#> xfun 0.45 2024-06-16 [1] RSPM (R 4.4.0)
#> XVector 0.44.0 2024-04-30 [1] Bioconductor 3.19 (R 4.4.1)
#> yaml 2.3.8 2023-12-11 [1] RSPM (R 4.4.0)
#> yulab.utils 0.1.4 2024-01-28 [1] RSPM (R 4.4.0)
#> zlibbioc 1.50.0 2024-04-30 [1] Bioconductor 3.19 (R 4.4.1)
#>
#> [1] /usr/local/lib/R/site-library
#> [2] /usr/local/lib/R/library
#>
#> ──────────────────────────────────────────────────────────────────────────────