lefser: a metagenomic biomarker discovery tool
Asya Khleborodova, Sehyun Oh, Ludwig Geistlinger, and Levi Waldron
2026-03-25
Source:vignettes/lefser.Rmd
lefser.RmdOverview
Background
lefser is the R implementation of the Linear discriminant analysis (LDA) Effect Size (LEfSe), a Python package for metagenomic biomarker discovery and explanation. (Huttenhower et al. 2011).
The original software utilizes standard statistical significance tests along with supplementary tests that incorporate biological consistency and the relevance of effects to identity the features (e.g., organisms, clades, OTU, genes, or functions) that are most likely to account for differences between the two sample classes of interest. While LEfSe is widely used and available in different platform such as Galaxy UI and Conda, there is no convenient way to incorporate it in R-based workflows. Thus, we re-implement LEfSe as an R/Bioconductor package, lefser. Following the LEfSe‘s algorithm including Kruskal-Wallis test, Wilcoxon-Rank Sum test, and Linear Discriminant Analysis, with some modifications, lefser successfully reproduces and improves the original statistical method and the associated plotting functionality.
Install and load pacakge
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("lefser")Citing lefser
Your citations are crucial in keeping our software free and open source. To cite our package, please use this publication at the link here.
Analysis example
Prepare input
lefser package include the demo dataset,
zeller14, which is the microbiome data from colorectal
cancer (CRC) patients and controls (Zeller
et al. 2014).
In this vignette, we excluded the ‘adenoma’ condition and used control/CRC as the main classes and age category as sub-classes (adult vs. senior) with different numbers of samples: control-adult (n = 46), control-senior (n = 20), CRC-adult (n = 45), and CRC-senior (n = 46).
data(zeller14)
zeller14 <- zeller14[, zeller14$study_condition != "adenoma"]The class and subclass information is stored in the
colData slot under the study_condition and
age_category columns, respectively.
## Contingency table
table(zeller14$age_category, zeller14$study_condition)#>
#> control CRC
#> adult 46 45
#> senior 20 46If you try to run lefser directly on the ‘zeller14’
data, you will get the following warning messages
lefser(zeller14, classCol = "study_condition", subclassCol = "age_category")Warning messages:
1: In lefser(zeller14, classCol = "study_condition", subclassCol = "age_category") :
Convert counts to relative abundances with 'relativeAb()'
2: In lda.default(x, classing, ...) : variables are collinearTerminal node
When working with taxonomic data, including both terminal and non-terminal nodes in the analysis can lead to collinearity problems. Non-terminal nodes (e.g., genus) are often linearly dependent on their corresponding terminal nodes (e.g., species) since the species-level information is essentially a subset or more specific representation of the genus-level information. This collinearity can violate the assumptions of certain statistical methods, such as linear discriminant analysis (LDA), and can lead to unstable or unreliable results. By using only terminal nodes, you can effectively eliminate this collinearity issue, ensuring that your analysis is not affected by linearly dependent or highly correlated variables. Additionally, you can benefit of avoiding redundancy, increasing specificity, simplifying data, and reducing ambiguity, using only terminal nodes.
You can select only the terminal node using
get_terminal_nodes function.
tn <- get_terminal_nodes(rownames(zeller14))
zeller14tn <- zeller14[tn,]Relative abundance
First warning message informs you that lefser requires
relative abundance of features. You can use relativeAb
function to reformat your input.
zeller14tn_ra <- relativeAb(zeller14tn)Run lefser
The lefser function returns a data.frame
with two columns - the names of selected features (the
features column) and their effect size (the
scores column).
There is a random number generation step in the lefser
algorithm to ensure that more than half of the values for each features
are unique. In most cases, inputs are sparse, so in practice, this step
is handling 0s. So to reproduce the identical result, you should set the
seed before running lefser.
set.seed(1234)
res <- lefser(zeller14tn_ra, # relative abundance only with terminal nodes
classCol = "study_condition",
subclassCol = "age_category")
head(res)#> features
#> 1 k__Bacteria|p__Firmicutes|c__Clostridia|o__Clostridiales|f__Ruminococcaceae|g__Ruminococcus|s__Ruminococcus_sp_5_1_39BFAA|t__GCF_000159975
#> 2 k__Bacteria|p__Firmicutes|c__Clostridia|o__Clostridiales|f__Eubacteriaceae|g__Eubacterium|s__Eubacterium_hallii|t__GCF_000173975
#> 3 k__Bacteria|p__Firmicutes|c__Bacilli|o__Lactobacillales|f__Streptococcaceae|g__Streptococcus|s__Streptococcus_salivarius|t__Streptococcus_salivarius_unclassified
#> 4 k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Bifidobacteriales|f__Bifidobacteriaceae|g__Bifidobacterium|s__Bifidobacterium_catenulatum|t__GCF_000173455
#> 5 k__Bacteria|p__Firmicutes|c__Clostridia|o__Clostridiales|f__Eubacteriaceae|g__Eubacterium|s__Eubacterium_ventriosum|t__GCF_000153885
#> 6 k__Bacteria|p__Firmicutes|c__Clostridia|o__Clostridiales|f__Lachnospiraceae|g__Lachnospiraceae_noname|s__Lachnospiraceae_bacterium_5_1_63FAA|t__GCF_000185525
#> scores
#> 1 -3.795313
#> 2 -3.700808
#> 3 -3.521229
#> 4 -3.154384
#> 5 -3.126024
#> 6 -3.082525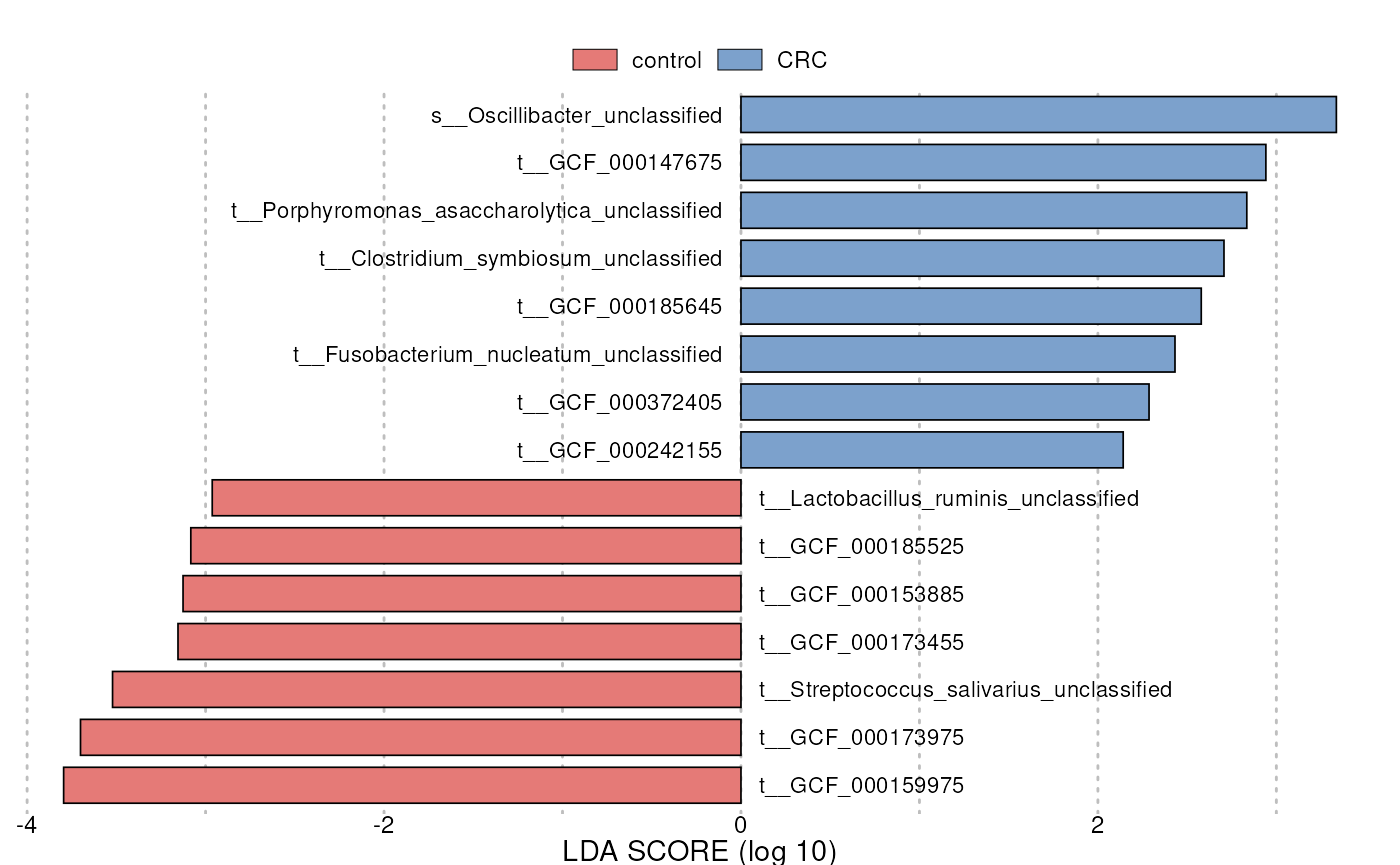
Benchmarking againt other tools
The codes for benchmarking lefser against LEfSe and the other R implementation of LEfSe is available here.
Interoperating with phyloseq
When using phyloseq objects, we recommend to extract the
data and create a SummarizedExperiment object as
follows:
library(phyloseq)
library(SummarizedExperiment)
## Load phyloseq object
fp <- system.file("extdata",
"study_1457_split_library_seqs_and_mapping.zip",
package = "phyloseq")
kostic <- microbio_me_qiime(fp)#> Found biom-format file, now parsing it...
#> Done parsing biom...
#> Importing Sample Metdadata from mapping file...
#> Merging the imported objects...
#> Successfully merged, phyloseq-class created.
#> Returning...
## Split data tables
counts <- unclass(otu_table(kostic))
coldata <- as(sample_data(kostic), "data.frame")
## Create a SummarizedExperiment object
SummarizedExperiment(assays = list(counts = counts), colData = coldata)#> class: SummarizedExperiment
#> dim: 2505 190
#> metadata(0):
#> assays(1): counts
#> rownames(2505): 304309 469478 ... 206906 298806
#> rowData names(0):
#> colnames(190): C0333.N.518126 C0333.T.518046 ... 32I9UNA9.518098
#> BFJMKNMP.518102
#> colData names(71): X.SampleID BarcodeSequence ... HOST_TAXID
#> DescriptionYou may also consider using
makeTreeSummarizedExperimentFromPhyloseq from the
mia package.
mia::makeTreeSummarizedExperimentFromPhyloseq(kostic)#> class: TreeSummarizedExperiment
#> dim: 2505 190
#> metadata(0):
#> assays(1): counts
#> rownames(2505): 304309 469478 ... 206906 298806
#> rowData names(7): Kingdom Phylum ... Genus Species
#> colnames(190): C0333.N.518126 C0333.T.518046 ... 32I9UNA9.518098
#> BFJMKNMP.518102
#> colData names(71): X.SampleID BarcodeSequence ... HOST_TAXID
#> Description
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):
#> rowLinks: NULL
#> rowTree: NULL
#> colLinks: NULL
#> colTree: NULLSession Info
#> R Under development (unstable) (2026-03-22 r89674)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: Etc/UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] phyloseq_1.55.2 lefser_1.20.2
#> [3] SummarizedExperiment_1.41.1 Biobase_2.71.0
#> [5] GenomicRanges_1.63.1 Seqinfo_1.1.0
#> [7] IRanges_2.45.0 S4Vectors_0.49.0
#> [9] BiocGenerics_0.57.0 generics_0.1.4
#> [11] MatrixGenerics_1.23.0 matrixStats_1.5.0
#> [13] BiocStyle_2.39.0
#>
#> loaded via a namespace (and not attached):
#> [1] libcoin_1.0-12 RColorBrewer_1.1-3
#> [3] jsonlite_2.0.0 MultiAssayExperiment_1.37.2
#> [5] magrittr_2.0.4 TH.data_1.1-5
#> [7] modeltools_0.2-24 ggbeeswarm_0.7.3
#> [9] farver_2.1.2 rmarkdown_2.30
#> [11] fs_2.0.1 ragg_1.5.2
#> [13] vctrs_0.7.2 multtest_2.67.0
#> [15] DelayedMatrixStats_1.33.0 ggtree_4.1.1
#> [17] htmltools_0.5.9 S4Arrays_1.11.1
#> [19] BiocNeighbors_2.5.4 SparseArray_1.11.11
#> [21] gridGraphics_0.5-1 sass_0.4.10
#> [23] bslib_0.10.0 htmlwidgets_1.6.4
#> [25] desc_1.4.3 DECIPHER_3.7.1
#> [27] plyr_1.8.9 sandwich_3.1-1
#> [29] testthat_3.3.2 zoo_1.8-15
#> [31] cachem_1.1.0 igraph_2.2.2
#> [33] lifecycle_1.0.5 iterators_1.0.14
#> [35] pkgconfig_2.0.3 rsvd_1.0.5
#> [37] Matrix_1.7-5 R6_2.6.1
#> [39] fastmap_1.2.0 digest_0.6.39
#> [41] aplot_0.2.9 patchwork_1.3.2
#> [43] scater_1.39.3 irlba_2.3.7
#> [45] textshaping_1.0.5 vegan_2.7-3
#> [47] beachmat_2.27.3 labeling_0.4.3
#> [49] TreeSummarizedExperiment_2.19.0 abind_1.4-8
#> [51] mgcv_1.9-4 compiler_4.6.0
#> [53] fontquiver_0.2.1 withr_3.0.2
#> [55] S7_0.2.1 BiocParallel_1.45.0
#> [57] DBI_1.3.0 viridis_0.6.5
#> [59] MASS_7.3-65 rappdirs_0.3.4
#> [61] DelayedArray_0.37.0 bluster_1.21.1
#> [63] biomformat_1.39.10 permute_0.9-10
#> [65] tools_4.6.0 vipor_0.4.7
#> [67] otel_0.2.0 beeswarm_0.4.0
#> [69] ape_5.8-1 glue_1.8.0
#> [71] nlme_3.1-168 grid_4.6.0
#> [73] mia_1.19.6 cluster_2.1.8.2
#> [75] reshape2_1.4.5 ade4_1.7-24
#> [77] gtable_0.3.6 tidyr_1.3.2
#> [79] data.table_1.18.2.1 BiocSingular_1.27.1
#> [81] ScaledMatrix_1.19.0 coin_1.4-3
#> [83] XVector_0.51.0 ggrepel_0.9.8
#> [85] foreach_1.5.2 pillar_1.11.1
#> [87] stringr_1.6.0 yulab.utils_0.2.4
#> [89] splines_4.6.0 dplyr_1.2.0
#> [91] treeio_1.35.0 lattice_0.22-9
#> [93] survival_3.8-6 DirichletMultinomial_1.53.0
#> [95] tidyselect_1.2.1 fontLiberation_0.1.0
#> [97] SingleCellExperiment_1.33.2 Biostrings_2.79.5
#> [99] scuttle_1.21.0 knitr_1.51
#> [101] fontBitstreamVera_0.1.1 gridExtra_2.3
#> [103] bookdown_0.46 xfun_0.57
#> [105] brio_1.1.5 stringi_1.8.7
#> [107] lazyeval_0.2.2 ggfun_0.2.0
#> [109] yaml_2.3.12 evaluate_1.0.5
#> [111] codetools_0.2-20 gdtools_0.5.0
#> [113] tibble_3.3.1 BiocManager_1.30.27
#> [115] ggplotify_0.1.3 cli_3.6.5
#> [117] systemfonts_1.3.2 jquerylib_0.1.4
#> [119] Rcpp_1.1.1 parallel_4.6.0
#> [121] pkgdown_2.2.0 ggplot2_4.0.2
#> [123] ecodive_2.2.2 sparseMatrixStats_1.23.0
#> [125] decontam_1.31.0 viridisLite_0.4.3
#> [127] mvtnorm_1.3-6 tidytree_0.4.7
#> [129] ggiraph_0.9.6 scales_1.4.0
#> [131] purrr_1.2.1 crayon_1.5.3
#> [133] rlang_1.1.7 multcomp_1.4-30