Analysis of prevalent stool genera in BugSigDB
05 May, 2025
healthysig.RmdBugSigDB stat- Signature overlaps
Compare genus abundance change (increased/decreased) from all diseased subjects in BugsigDB with existing typical genus from healthy volunteers recorded in BugsigDB Study 562 typical genus were set at prevalence level=0, 50, and 70 Using Feces sample as an example
Get typical healthy signatures from Study 562
##
## Attaching package: 'dplyr'## The following objects are masked from 'package:stats':
##
## filter, lag## The following objects are masked from 'package:base':
##
## intersect, setdiff, setequal, union## Loading required package: ggplot2
# set light theme for ggplot
theme_set(theme_light())
full.dat <- bugsigdbr::importBugSigDB(cache=FALSE)
#Stripping empty signatures:
ind1 <- lengths(full.dat[["MetaPhlAn taxon names"]]) > 0
ind2 <- lengths(full.dat[["NCBI Taxonomy IDs"]]) > 0
dat <- full.dat[ind1 & ind2,]
#prevalence threshold=50%
bugs50<- (dat) %>%
filter (Study=='Study 562') %>%
filter (Experiment=="Experiment 3" & `Signature page name`=='Signature 1') %>%
filter (`Body site` == "Feces") %>%
filter (grepl("genus",Description))
healthysig50 <- bugsigdbr::getSignatures(bugs50)
head(healthysig50)## $`bsdb:562/3/1_Health-study-participation:feces-from-healthy-adult_vs_none---50%-prevalence-threshold_NA`
## [1] "447020" "2048137" "239934" "239759" "2569097" "207244" "816"
## [8] "397864" "1678" "35832" "572511" "574697" "1485" "102106"
## [15] "33042" "189330" "561" "1730" "216851" "946234" "1407607"
## [22] "204475" "644652" "1505657" "28050" "2172004" "2316020" "283168"
## [29] "459786" "375288" "577310" "33024" "909656" "838" "841"
## [36] "1263" "1905344" "1301"this code didn’t work because experiment 10 has no signatures
#prevalence threshold=0
bugs0<- (dat) %>%
filter (Study=='Study 562') %>%
filter (Experiment=="Experiment 10") %>%
filter (`Body site` == "Feces") %>%
filter (grepl("genus",Description))
healthysig0 <- bugsigdbr::getSignatures(bugs0)
head(healthysig0)
#prevalence threshold=70%
bugs70<- (dat) %>%
filter (Study=='Study 562') %>%
filter (Experiment=="Experiment 9" & `Signature page name`=='Signature 1') %>%
filter (`Body site` == "Feces") %>%
filter (grepl("genus",Description))
healthysig70 <- bugsigdbr::getSignatures(bugs70)
head(healthysig70)## $`bsdb:562/9/1_Health-study-participation:feces-from-healthy-adult_vs_none---70%-prevalence-threshold_NA`
## [1] "447020" "2048137" "239759" "2569097" "207244" "816" "1678"
## [8] "572511" "1485" "102106" "33042" "189330" "1730" "216851"
## [15] "946234" "1407607" "204475" "28050" "2316020" "283168" "459786"
## [22] "375288" "909656" "841" "1263" "1301" "1905344"Get changed in abundance disease genus
bugdisease_increased <- dat %>%
filter (`Host species` == 'Homo sapiens') %>%
filter (`Body site` == 'Feces') %>%
filter (`Abundance in Group 1`=="increased")
#get genus level sig change in abundance only
diseasesig_increased <- bugsigdbr::getSignatures(bugdisease_increased, tax.level='genus')
length(diseasesig_increased)## [1] 1390
regexp <- "[[:digit:]]+"
bugdisease_increased2<-bugdisease_increased%>%mutate(
study_id=stringr::str_extract(Study, regexp),
experiment_id=stringr::str_extract(Experiment, regexp),
signature_id=stringr::str_extract(`Signature page name`, regexp)
)
diseasesig_increased2 <- purrr::map_df(diseasesig_increased, ~as.data.frame(.x), .id="id") %>%
# tidyr::pivot_wider(id_cols=id) %>%
mutate(name2_1=stringr::str_replace(id, "bsdb:", "")) %>%
mutate(name2_2=(stringr::str_split(name2_1,"/"))) %>%
mutate(study_id= purrr::map_chr(name2_2, 1),
experiment_id= purrr::map_chr(name2_2, 2),
signature_id= purrr::map_chr(stringr::str_split(purrr::map_chr(name2_2, 3),'_'),1)
) %>%
dplyr::select(-c("name2_1","name2_2"))
#exclude studies does not return genus level abundance change
bugall_increased <- merge(x = bugdisease_increased2, y = diseasesig_increased2,
by =c('study_id','experiment_id','signature_id'), all.x = TRUE)
bugdisease_decreased <- dat %>%
filter (`Host species` == 'Homo sapiens') %>%
filter (`Body site` == 'Feces') %>%
filter (`Abundance in Group 1`=="decreased")
#get genus level sig change in abundance only
diseasesig_decreased <- bugsigdbr::getSignatures(bugdisease_decreased, tax.level='genus')
length(diseasesig_decreased)## [1] 1315
regexp <- "[[:digit:]]+"
bugdisease_decreased2<-bugdisease_decreased%>%mutate(
study_id=stringr::str_extract(Study, regexp),
experiment_id=stringr::str_extract(Experiment, regexp),
signature_id=stringr::str_extract(`Signature page name`, regexp)
)
diseasesig_decreased2 <- purrr::map_df(diseasesig_decreased, ~as.data.frame(.x), .id="id") %>%
# tidyr::pivot_wider(id_cols=id) %>%
mutate(name2_1=stringr::str_replace(id, "bsdb:", "")) %>%
mutate(name2_2=(stringr::str_split(name2_1,"/"))) %>%
mutate(study_id= purrr::map_chr(name2_2, 1),
experiment_id= purrr::map_chr(name2_2, 2),
signature_id= purrr::map_chr(stringr::str_split(purrr::map_chr(name2_2, 3),'_'),1)
) %>%
dplyr::select(-c("name2_1","name2_2"))
#exclude studies does not return genus level abundance change
bugall_decreased <- merge(x = diseasesig_decreased2, y = bugdisease_decreased2,
by =c('study_id','experiment_id','signature_id'), all.x = TRUE)Calculate pairwise overlaps
#this code didn’t work because experiment 10 has no signatures,
therefore line 130 - 180 were commented out
{r overlap_prevelance=0} #{r overlap_prevelance=0}
experiment 10 has no signatures
library(purrr) list0_decreased <- c(healthysig0, diseasesig_decreased) paircomp0_decreased <- calcPairwiseOverlaps(list0_decreased)
paircheck0_decreased <- paircomp0_decreased %>% filter(stringr::str_detect(name1, ‘feces-from-healthy-adult_vs_none—0%-prevalence-threshold’)) %>% mutate(name2_1=stringr::str_replace(name2, “bsdb:”, ““)) %>% mutate(name2_2=(stringr::str_split(name2_1,”/“))) %>% mutate(study_id= purrr::map_chr(name2_2, 1), experiment_id= purrr::map_chr(name2_2, 2), signature_id= purrr::map_chr(stringr::str_split(purrr::map_chr(name2_2, 3),’_’),1) )
list0_increased <- c(healthysig0, diseasesig_increased) paircomp0_increased <- calcPairwiseOverlaps(list0_increased)
paircheck0_increased <- paircomp0_increased %>% filter(stringr::str_detect(name1, ‘feces-from-healthy-adult_vs_none—0%-prevalence-threshold’)) %>% mutate(name2_1=stringr::str_replace(name2, “bsdb:”, ““)) %>% mutate(name2_2=(stringr::str_split(name2_1,”/“))) %>% mutate(study_id= purrr::map_chr(name2_2, 1), experiment_id= purrr::map_chr(name2_2, 2), signature_id= purrr::map_chr(stringr::str_split(purrr::map_chr(name2_2, 3),’_’),1) )
# merge jacard scores with disease study data
The issue why I need to merge overlap score back with all disease budsigdb studies is that calcPairwiseOverlaps drops all 0 intersect pairs
#```{r merge overlap scores with all disease study data}
overlap0_decreased<- merge(x = bugall_decreased, y = paircheck0_decreased, by =c('study_id','experiment_id','signature_id'), all.x = TRUE) %>%
mutate(type="decreased")%>%
filter(length_set2>4) #keep only bugsigdb studies that tested more than 4 increased or decreased
overlap0_decreased[is.na(overlap0_decreased)] <- 0
overlap0_increased<- merge(x = bugall_increased, y = paircheck0_increased, by =c('study_id','experiment_id','signature_id'), all.x = TRUE) %>%
mutate(type="increased") %>%
filter(length_set2 > 4) #keep only bugsigdb studies that tested more than 4 increased or decreased
overlap0_increased[is.na(overlap0_increased)] <- 0 #{r comparison-increased_vs_decrease}
t.test(overlap0_increasedoverlap)
wilcox.test(overlap0_increasedoverlap)
p <- rbind(overlap0_increased,overlap0_decreased) %>% ggplot( aes(x=overlap, fill=type)) + geom_histogram( color=“#e9ecef”, alpha=0.6, position = ‘dodge’,bins=20) p
``` r
library(purrr)
list50_decreased <- c(healthysig50, diseasesig_decreased)
paircomp50_decreased <- calcPairwiseOverlaps(list50_decreased)
paircheck50_decreased <- paircomp50_decreased %>%
filter(stringr::str_detect(name1, 'feces-from-healthy-adult_vs_none---50%-prevalence-threshold')) %>%
mutate(name2_1=stringr::str_replace(name2, "bsdb:", "")) %>%
mutate(name2_2=(stringr::str_split(name2_1,"/"))) %>%
mutate(study_id= purrr::map_chr(name2_2, 1),
experiment_id= purrr::map_chr(name2_2, 2),
signature_id= purrr::map_chr(stringr::str_split(purrr::map_chr(name2_2, 3),'_'),1)
)
list50_increased <- c(healthysig50, diseasesig_increased)
paircomp50_increased <- calcPairwiseOverlaps(list50_increased)
paircheck50_increased <- paircomp50_increased %>%
filter(stringr::str_detect(name1, 'feces-from-healthy-adult_vs_none---50%-prevalence-threshold')) %>%
mutate(name2_1=stringr::str_replace(name2, "bsdb:", "")) %>%
mutate(name2_2=(stringr::str_split(name2_1,"/"))) %>%
mutate(study_id= purrr::map_chr(name2_2, 1),
experiment_id= purrr::map_chr(name2_2, 2),
signature_id= purrr::map_chr(stringr::str_split(purrr::map_chr(name2_2, 3),'_'),1)
) merge jacard scores with disease study data
overlap50_decreased<- merge(x = bugall_decreased, y = paircheck50_decreased, by =c('study_id','experiment_id','signature_id'), all.x = TRUE) %>%
mutate(type="decreased")%>%
filter(length_set2>4) #keep only bugsigdb studies that tested more than 4 increased or decreased
overlap50_decreased[is.na(overlap50_decreased)] <- 0
overlap50_increased<- merge(x = bugall_increased, y = paircheck50_increased, by =c('study_id','experiment_id','signature_id'), all.x = TRUE) %>%
mutate(type="increased") %>%
filter(length_set2 > 4) #keep only bugsigdb studies that tested more than 4 increased or decreased
overlap50_increased[is.na(overlap50_increased)] <- 0
t.test(overlap50_increased$overlap, overlap50_decreased$overlap)##
## Welch Two Sample t-test
##
## data: overlap50_increased$overlap and overlap50_decreased$overlap
## t = -18.365, df = 8268.9, p-value < 2.2e-16
## alternative hypothesis: true difference in means is not equal to 0
## 95 percent confidence interval:
## -0.09618345 -0.07763084
## sample estimates:
## mean of x mean of y
## 0.3960865 0.4829937
wilcox.test(overlap50_increased$overlap, overlap50_decreased$overlap)##
## Wilcoxon rank sum test with continuity correction
##
## data: overlap50_increased$overlap and overlap50_decreased$overlap
## W = 6665081, p-value < 2.2e-16
## alternative hypothesis: true location shift is not equal to 0
p <- rbind(overlap50_increased,overlap50_decreased) %>%
ggplot( aes(x=overlap, fill=type)) +
geom_histogram( color="#e9ecef", alpha=0.6, position = 'dodge',bins=20)
p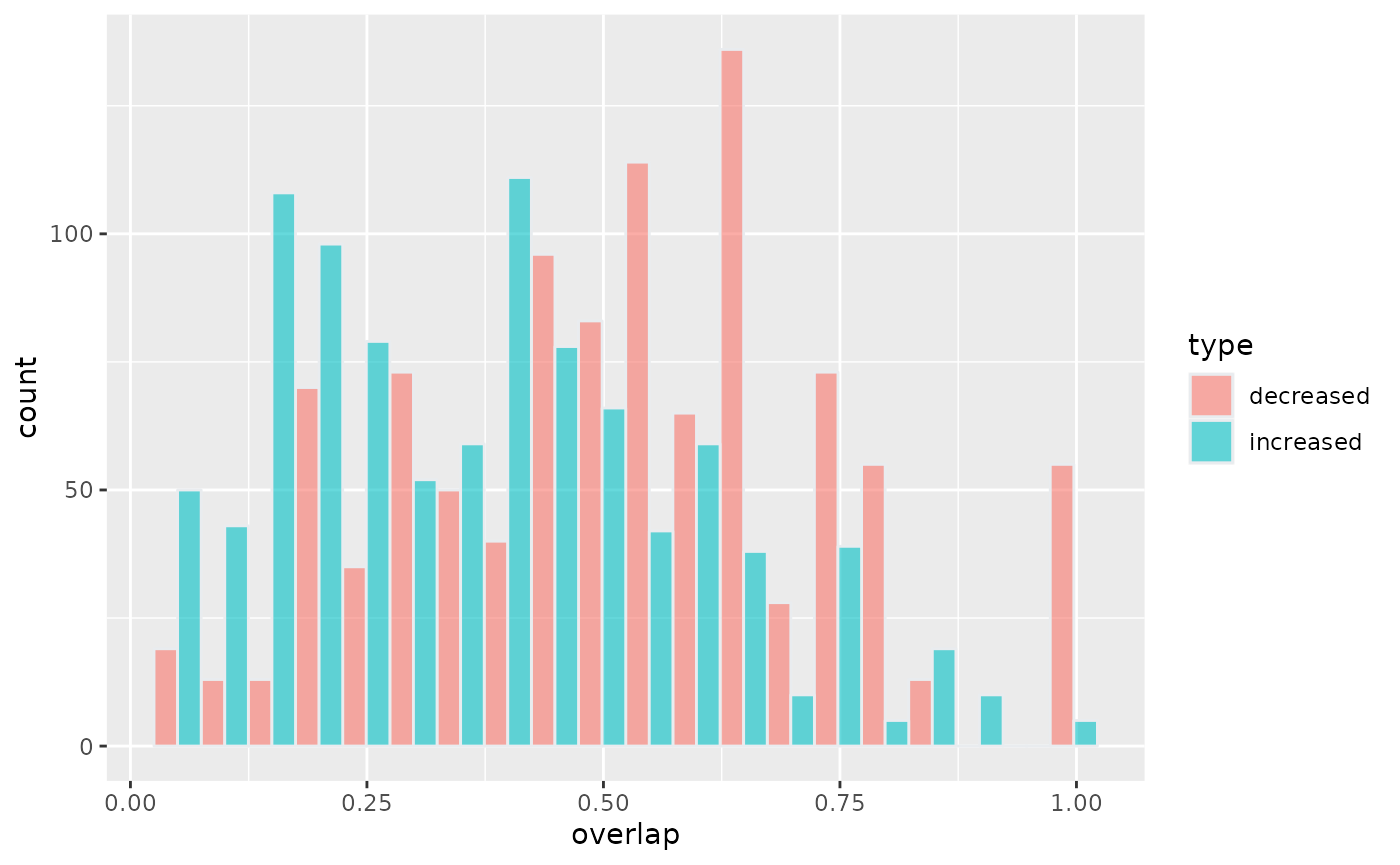
library(purrr)
list70_decreased <- c(healthysig70, diseasesig_decreased)
paircomp70_decreased <- calcPairwiseOverlaps(list70_decreased)
paircheck70_decreased <- paircomp70_decreased %>%
filter(stringr::str_detect(name1, 'feces-from-healthy-adult_vs_none---70%-prevalence-threshold')) %>%
mutate(name2_1=stringr::str_replace(name2, "bsdb:", "")) %>%
mutate(name2_2=(stringr::str_split(name2_1,"/"))) %>%
mutate(study_id= purrr::map_chr(name2_2, 1),
experiment_id= purrr::map_chr(name2_2, 2),
signature_id= purrr::map_chr(stringr::str_split(purrr::map_chr(name2_2, 3),'_'),1)
)
list70_increased <- c(healthysig70, diseasesig_increased)
paircomp70_increased <- calcPairwiseOverlaps(list70_increased)
paircheck70_increased <- paircomp70_increased %>%
filter(stringr::str_detect(name1, 'feces-from-healthy-adult_vs_none---70%-prevalence-threshold')) %>%
mutate(name2_1=stringr::str_replace(name2, "bsdb:", "")) %>%
mutate(name2_2=(stringr::str_split(name2_1,"/"))) %>%
mutate(study_id= purrr::map_chr(name2_2, 1),
experiment_id= purrr::map_chr(name2_2, 2),
signature_id= purrr::map_chr(stringr::str_split(purrr::map_chr(name2_2, 3),'_'),1)
) merge jacard scores with disease study data
overlap70_decreased<- merge(x = bugall_decreased, y = paircheck70_decreased, by =c('study_id','experiment_id','signature_id'), all.x = TRUE) %>%
mutate(type="decreased")%>%
filter(length_set2>4) #keep only bugsigdb studies that tested more than 4 increased or decreased
overlap70_decreased[is.na(overlap70_decreased)] <- 0
overlap70_increased<- merge(x = bugall_increased, y = paircheck70_increased, by =c('study_id','experiment_id','signature_id'), all.x = TRUE) %>%
mutate(type="increased") %>%
filter(length_set2 > 4) #keep only bugsigdb studies that tested more than 4 increased or decreased
overlap70_increased[is.na(overlap70_increased)] <- 0
t.test(overlap70_increased$overlap, overlap70_decreased$overlap)
wilcox.test(overlap70_increased$overlap, overlap70_decreased$overlap)
p <- rbind(overlap70_increased,overlap70_decreased) %>%
ggplot( aes(x=overlap, fill=type)) +
geom_histogram( color="#e9ecef", alpha=0.6, position = 'dodge',bins=20)
p