cBioPortalData: User Guide
Marcel Ramos & Levi Waldron
March 25, 2026
Source:vignettes/cBioPortalData.Rmd
cBioPortalData.RmdIntroduction
The cBioPortal for Cancer Genomics website is a great resource for interactive exploration of study datasets. However, it does not easily allow the analyst to obtain and further analyze the data.
We’ve developed the cBioPortalData package to fill this
need to programmatically access the data resources available on the
cBioPortal.
The cBioPortalData package provides an R interface for
accessing the cBioPortal study data within the Bioconductor
ecosystem.
It downloads study data from the cBioPortal API (the full API specification can be found here https://cbioportal.org/api) and uses Bioconductor infrastructure to cache and represent the data.
We demonstrate common use cases of cBioPortalData and
curatedTCGAData during Bioconductor conference workshops.
We use the MultiAssayExperiment (Ramos et al. 2017) package to integrate,
represent, and coordinate multiple experiments for the studies available
in the cBioPortal. This package in conjunction with
curatedTCGAData give access to a large trove of publicly
available bioinformatic data. Please see our JCO Clinical Cancer
Informatics publication (Ramos et al.
2020).
Citations
Our free and open source project depends on citations for funding.
When using cBioPortalData, please cite the following
publications:
Overview
Data Structures
Data are provided as a single MultiAssayExperiment per
study. The MultiAssayExperiment representation usually
contains SummarizedExperiment objects for expression data
and RaggedExperiment objects for mutation and CNV-type
data. RaggedExperiment is a data class for representing
‘ragged’ genomic location data, meaning that the measurements per sample
vary.
For more information, please see the RaggedExperiment
and SummarizedExperiment vignettes.
Identifying available studies
As we work through the data, there are some datasest that cannot be
represented as MultiAssayExperiment objects. This can be
due to a number of reasons such as the way the data is handled, presence
of mis-matched identifiers, invalid data types, etc. To see what
datasets are currently not building, we can look refer to
getStudies() with the buildReport = TRUE
argument.
cbio <- cBioPortal()
studies <- getStudies(cbio, buildReport = TRUE)
head(studies)## # A tibble: 6 × 15
## name description publicStudy pmid citation groups status importDate
## <chr> <chr> <lgl> <chr> <chr> <chr> <int> <chr>
## 1 Mixed cfDNA (… "IMPACT se… TRUE 3405… Tsui et… "TSUI… 0 2026-01-0…
## 2 Uterine Endom… "CPTAC Ute… TRUE NA NA "PUBL… 0 2026-01-1…
## 3 Mantle Cell L… "Whole exo… TRUE 2414… Beà et… "" 0 2026-01-0…
## 4 Anaplastic Th… "Whole-gen… TRUE 3841… Zeng, P… "" 0 2026-01-1…
## 5 Stomach Adeno… "Whole gen… TRUE 2481… Wang et… "PUBL… 0 2026-01-1…
## 6 Non-Small Cel… "Whole exo… TRUE 2576… Rivzi e… "PUBL… 0 2026-01-0…
## # ℹ 7 more variables: allSampleCount <int>, readPermission <lgl>,
## # studyId <chr>, cancerTypeId <chr>, referenceGenome <chr>, api_build <lgl>,
## # pack_build <lgl>The last two columns will show the availability of each
studyId for either download method (pack_build
for cBioDataPack and api_build for
cBioPortalData).
Choosing download method
There are two main user-facing functions for downloading data from the cBioPortal API.
cBioDataPackmakes use of the tarball distribution of study data. This is useful when the user wants to download and analyze the entirety of the data as available from the cBioPortal.org website.cBioPortalDataallows a more flexibile approach to obtaining study data based on the available parameters such as molecular profile identifiers. This option is useful for users who have a set of gene symbols or identifiers and would like to get a smaller subset of the data that correspond to a particular molecular profile.
Two main functions
cBioDataPack: Obtain Study Data as Zipped Tarballs
This function will access the packaged data from https://cbioportal.org/datasets and return an
integrative MultiAssayExperiment representation.
## Use ask=FALSE for non-interactive use
laml <- cBioDataPack("laml_tcga", ask = FALSE)
laml## A MultiAssayExperiment object of 12 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 12:
## [1] cna_hg19.seg: RaggedExperiment with 13571 rows and 191 columns
## [2] cna: SummarizedExperiment with 24776 rows and 191 columns
## [3] linear_cna: SummarizedExperiment with 24776 rows and 191 columns
## [4] methylation_hm27: SummarizedExperiment with 10968 rows and 194 columns
## [5] methylation_hm450: SummarizedExperiment with 10968 rows and 194 columns
## [6] mrna_seq_rpkm_zscores_ref_all_samples: SummarizedExperiment with 19720 rows and 179 columns
## [7] mrna_seq_rpkm_zscores_ref_diploid_samples: SummarizedExperiment with 19719 rows and 179 columns
## [8] mrna_seq_rpkm: SummarizedExperiment with 19720 rows and 179 columns
## [9] mrna_seq_v2_rsem_zscores_ref_all_samples: SummarizedExperiment with 20531 rows and 173 columns
## [10] mrna_seq_v2_rsem_zscores_ref_diploid_samples: SummarizedExperiment with 20440 rows and 173 columns
## [11] mrna_seq_v2_rsem: SummarizedExperiment with 20531 rows and 173 columns
## [12] mutations: RaggedExperiment with 2584 rows and 197 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filescBioPortalData: Obtain data from the cBioPortal API
This function provides a more flexible and granular way to request a
MultiAssayExperiment object from a study ID, molecular
profile, set of genes, and a sample list.
In this example, we will obtain data from the Adrenocortical
carcinoma (ACC; acc_tcga) study. The list of
genes below are based on potential driving alterations
including amplifications, deletions, and point mutations. Also included
are a set of genes known to initiate familial syndromes including
adrenocortical neoplasms as described in Zheng et
al. (2016).
acc <- cBioPortalData(
api = cbio,
by = "hugoGeneSymbol",
studyId = "acc_tcga",
genes = c(
"TERT", "TERF2", "CDK4", "ZNRF3", "CDKN2A", "RB1", "RPL22",
"TP53", "CTNNB1", "PRKAR1A", "MEN1"
),
molecularProfileIds = c("acc_tcga_linear_CNA", "acc_tcga_mutations"),
)## harmonizing input:
## removing 1 colData rownames not in sampleMap 'primary'
acc## A MultiAssayExperiment object of 2 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 2:
## [1] acc_tcga_mutations: RangedSummarizedExperiment with 54 rows and 37 columns
## [2] acc_tcga_linear_CNA: SummarizedExperiment with 11 rows and 90 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save data to flat filesNote. To avoid overloading the API service, the API was designed to only query a part of the study data. Therefore, the user is required to enter either a set of genes of interest or a gene panel identifier.
Considerations
Note that cBioPortalData and cBioDataPack
obtain data diligently curated by the cBio Portal data team. The
original data and curation lies in the https://github.com/cBioPortal/cBioPortal GitHub
repository. However, despite the curation efforts there may be some
inconsistencies in identifiers in the data. This causes our software to
not work as intended though we have made efforts to represent all the
data from both API and tarball formats.
metadata
You may notice that the metadata() may have some
additional data that was not able to be integrated in the
MultiAssayExperiment.
metadata(acc)## [[1]]
## # A tibble: 62 × 22
## uniqueSampleKey uniquePatientKey molecularProfileId patientId entrezGeneId
## <chr> <chr> <chr> <chr> <int>
## 1 VENHQS1PUi1BNUoyL… VENHQS1PUi1BNUo… acc_tcga_mutations TCGA-OR-… 1499
## 2 VENHQS1PUi1BNUoyL… VENHQS1PUi1BNUo… acc_tcga_mutations TCGA-OR-… 7157
## 3 VENHQS1PUi1BNUozL… VENHQS1PUi1BNUo… acc_tcga_mutations TCGA-OR-… 5573
## 4 VENHQS1PUi1BNUo1L… VENHQS1PUi1BNUo… acc_tcga_mutations TCGA-OR-… 4221
## 5 VENHQS1PUi1BNUo1L… VENHQS1PUi1BNUo… acc_tcga_mutations TCGA-OR-… 6146
## 6 VENHQS1PUi1BNUo1L… VENHQS1PUi1BNUo… acc_tcga_mutations TCGA-OR-… 7157
## 7 VENHQS1PUi1BNUo1L… VENHQS1PUi1BNUo… acc_tcga_mutations TCGA-OR-… 84133
## 8 VENHQS1PUi1BNUo4L… VENHQS1PUi1BNUo… acc_tcga_mutations TCGA-OR-… 7157
## 9 VENHQS1PUi1BNUpBL… VENHQS1PUi1BNUp… acc_tcga_mutations TCGA-OR-… 4221
## 10 VENHQS1PUi1BNUpBL… VENHQS1PUi1BNUp… acc_tcga_mutations TCGA-OR-… 5573
## # ℹ 52 more rows
## # ℹ 17 more variables: studyId <chr>, center <chr>, mutationStatus <chr>,
## # validationStatus <chr>, tumorAltCount <int>, tumorRefCount <int>,
## # normalAltCount <int>, normalRefCount <int>, referenceAllele <chr>,
## # proteinChange <chr>, variantType <chr>, keyword <chr>, variantAllele <chr>,
## # refseqMrnaId <chr>, proteinPosStart <int>, proteinPosEnd <int>, type <chr>
##
## [[2]]
## # A tibble: 990 × 7
## uniqueSampleKey uniquePatientKey entrezGeneId molecularProfileId patientId
## <chr> <chr> <int> <chr> <chr>
## 1 VENHQS1PUi1BNUoxL… VENHQS1PUi1BNUo… 4221 acc_tcga_linear_C… TCGA-OR-…
## 2 VENHQS1PUi1BNUoxL… VENHQS1PUi1BNUo… 5925 acc_tcga_linear_C… TCGA-OR-…
## 3 VENHQS1PUi1BNUoxL… VENHQS1PUi1BNUo… 6146 acc_tcga_linear_C… TCGA-OR-…
## 4 VENHQS1PUi1BNUoxL… VENHQS1PUi1BNUo… 84133 acc_tcga_linear_C… TCGA-OR-…
## 5 VENHQS1PUi1BNUoxL… VENHQS1PUi1BNUo… 1019 acc_tcga_linear_C… TCGA-OR-…
## 6 VENHQS1PUi1BNUoxL… VENHQS1PUi1BNUo… 1029 acc_tcga_linear_C… TCGA-OR-…
## 7 VENHQS1PUi1BNUoxL… VENHQS1PUi1BNUo… 7014 acc_tcga_linear_C… TCGA-OR-…
## 8 VENHQS1PUi1BNUoxL… VENHQS1PUi1BNUo… 7015 acc_tcga_linear_C… TCGA-OR-…
## 9 VENHQS1PUi1BNUoxL… VENHQS1PUi1BNUo… 7157 acc_tcga_linear_C… TCGA-OR-…
## 10 VENHQS1PUi1BNUoxL… VENHQS1PUi1BNUo… 1499 acc_tcga_linear_C… TCGA-OR-…
## # ℹ 980 more rows
## # ℹ 2 more variables: studyId <chr>, type <chr>Build prompts
You will also get a message for studyIds whose data has
not been fully integrated into a MultiAssayExperiment.
## Our testing shows that '%s' is not currently building.
## Use 'downloadStudy()' to manually obtain the data.
## Proceed anyway? [y/n]: yManual downloads
For this reason, we have also provided the
downloadStudy, untarStudy, and
loadStudy functions to allow researchers to simply download
the data and potentially, manually curate it. Generally, we advise
researchers to report inconsistencies in the data in the cBioPortal data repository.
Clearing the cache
cBioDataPack
In cases where a download is interrupted, the user may experience a
corrupt cache. The user can clear the cache for a particular study by
using the removeCache function. Note that this function
only works for data downloaded through the cBioDataPack
function.
removeCache("laml_tcga")cBioPortalData
For users who wish to clear the entire cBioPortalData
cache, it is recommended that they use:
unlink("~/.cache/cBioPortalData/")Example Analysis: Kaplan-Meier Plot
We can use information in the colData to draw a K-M plot
with a few variables from the colData slot of the
MultiAssayExperiment. First, we load the necessary
packages:
We can check the data to lookout for any issues.
table(colData(laml)$OS_STATUS)##
## 0:LIVING 1:DECEASED
## 67 133
class(colData(laml)$OS_MONTHS)## [1] "character"Now, we clean the data a bit to ensure that our variables are of the right type for the subsequent survival model fit.
collaml <- colData(laml)
collaml[collaml$OS_MONTHS == "[Not Available]", "OS_MONTHS"] <- NA
collaml$OS_MONTHS <- as.numeric(collaml$OS_MONTHS)
colData(laml) <- collamlWe specify a simple survival model using SEX as a
covariate and we draw the K-M plot.
fit <- survfit(
Surv(OS_MONTHS, as.numeric(substr(OS_STATUS, 1, 1))) ~ SEX,
data = colData(laml)
)
ggsurvplot(fit, data = colData(laml), risk.table = TRUE)## Ignoring unknown labels:
## • colour : "Strata"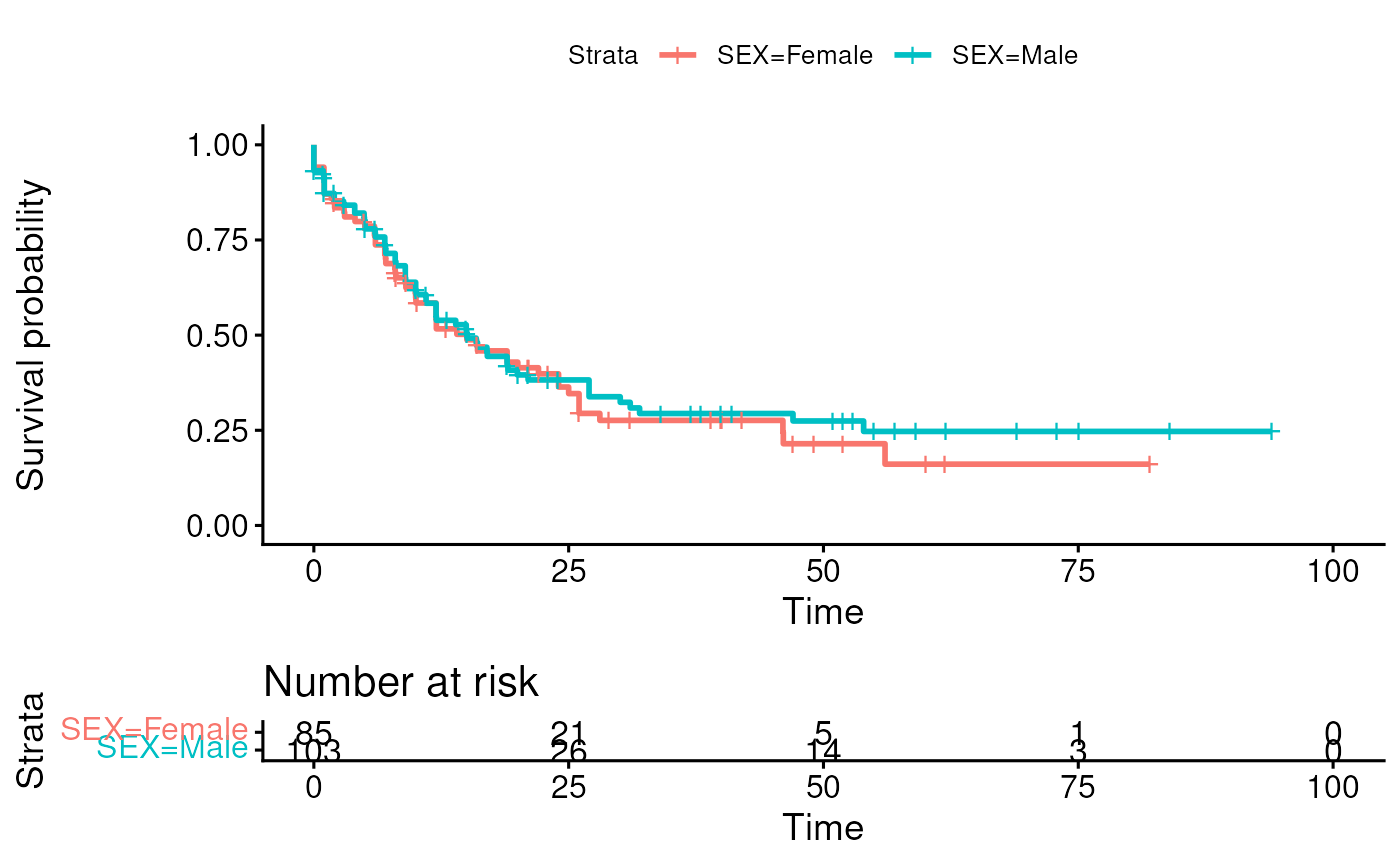
Data update requests
If you are interested in a particular study dataset that is not currently building, please open an issue at our GitHub repository and we will do our best to resolve the issues with the code base. Data issues can be opened at the cBioPortal data repository.
We appreciate your feedback!
sessionInfo
Click to see session info
## R Under development (unstable) (2026-03-22 r89674)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: Etc/UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] survminer_0.5.2 ggpubr_0.6.3
## [3] ggplot2_4.0.2 survival_3.8-6
## [5] cBioPortalData_2.22.3 MultiAssayExperiment_1.37.2
## [7] SummarizedExperiment_1.41.1 Biobase_2.71.0
## [9] GenomicRanges_1.63.1 Seqinfo_1.1.0
## [11] IRanges_2.45.0 S4Vectors_0.49.0
## [13] BiocGenerics_0.57.0 generics_0.1.4
## [15] MatrixGenerics_1.23.0 matrixStats_1.5.0
## [17] AnVIL_1.23.8 AnVILBase_1.5.1
## [19] dplyr_1.2.0 BiocStyle_2.39.0
##
## loaded via a namespace (and not attached):
## [1] RColorBrewer_1.1-3 jsonlite_2.0.0
## [3] magrittr_2.0.4 GenomicFeatures_1.63.1
## [5] farver_2.1.2 rmarkdown_2.30
## [7] fs_2.0.1 BiocIO_1.21.0
## [9] ragg_1.5.2 vctrs_0.7.2
## [11] memoise_2.0.1 Rsamtools_2.27.1
## [13] RCurl_1.98-1.18 rstatix_0.7.3
## [15] htmltools_0.5.9 S4Arrays_1.11.1
## [17] BiocBaseUtils_1.13.0 lambda.r_1.2.4
## [19] curl_7.0.0 broom_1.0.12
## [21] Formula_1.2-5 SparseArray_1.11.11
## [23] sass_0.4.10 bslib_0.10.0
## [25] htmlwidgets_1.6.4 desc_1.4.3
## [27] httr2_1.2.2 futile.options_1.0.1
## [29] cachem_1.1.0 commonmark_2.0.0
## [31] GenomicAlignments_1.47.0 mime_0.13
## [33] lifecycle_1.0.5 pkgconfig_2.0.3
## [35] Matrix_1.7-5 R6_2.6.1
## [37] fastmap_1.2.0 shiny_1.13.0
## [39] digest_0.6.39 GCPtools_1.1.0
## [41] RaggedExperiment_1.35.0 AnnotationDbi_1.73.0
## [43] textshaping_1.0.5 RSQLite_2.4.6
## [45] labeling_0.4.3 filelock_1.0.3
## [47] RTCGAToolbox_2.41.0 RJSONIO_2.0.0
## [49] httr_1.4.8 abind_1.4-8
## [51] compiler_4.6.0 bit64_4.6.0-1
## [53] withr_3.0.2 backports_1.5.0
## [55] S7_0.2.1 BiocParallel_1.45.0
## [57] carData_3.0-6 DBI_1.3.0
## [59] ggsignif_0.6.4 rappdirs_0.3.4
## [61] DelayedArray_0.37.0 rjson_0.2.23
## [63] tools_4.6.0 otel_0.2.0
## [65] httpuv_1.6.17 glue_1.8.0
## [67] restfulr_0.0.16 promises_1.5.0
## [69] gridtext_0.1.6 grid_4.6.0
## [71] gtable_0.3.6 tzdb_0.5.0
## [73] tidyr_1.3.2 data.table_1.18.2.1
## [75] hms_1.1.4 car_3.1-5
## [77] xml2_1.5.2 utf8_1.2.6
## [79] XVector_0.51.0 markdown_2.0
## [81] pillar_1.11.1 stringr_1.6.0
## [83] later_1.4.8 splines_4.6.0
## [85] ggtext_0.1.2 BiocFileCache_3.1.0
## [87] lattice_0.22-9 rtracklayer_1.71.3
## [89] bit_4.6.0 tidyselect_1.2.1
## [91] Biostrings_2.79.5 miniUI_0.1.2
## [93] knitr_1.51 gridExtra_2.3
## [95] litedown_0.9 bookdown_0.46
## [97] futile.logger_1.4.9 xfun_0.57
## [99] DT_0.34.0 stringi_1.8.7
## [101] UCSC.utils_1.7.1 yaml_2.3.12
## [103] evaluate_1.0.5 codetools_0.2-20
## [105] cigarillo_1.1.0 tibble_3.3.1
## [107] BiocManager_1.30.27 cli_3.6.5
## [109] xtable_1.8-8 systemfonts_1.3.2
## [111] jquerylib_0.1.4 Rcpp_1.1.1
## [113] GenomeInfoDb_1.47.2 GenomicDataCommons_1.35.1
## [115] dbplyr_2.5.2 png_0.1-9
## [117] XML_3.99-0.23 rapiclient_0.1.8
## [119] parallel_4.6.0 TCGAutils_1.31.4
## [121] pkgdown_2.2.0 readr_2.2.0
## [123] blob_1.3.0 bitops_1.0-9
## [125] scales_1.4.0 purrr_1.2.1
## [127] crayon_1.5.3 rlang_1.1.7
## [129] KEGGREST_1.51.1 rvest_1.0.5
## [131] formatR_1.14