NUI Galway Metagenomics Workshop
Lucas Schiffer, MPH
Section of Computational Biomedicine, Boston University School of Medicine, Boston, MA, U.S.A.schifferl@bu.edu
October 6, 2021
Source:vignettes/NUI-Galway-Metagenomics-Workshop.Rmd
NUI-Galway-Metagenomics-Workshop.RmdR Packages for This Tutorial
This tutorial demonstrates basic usage of curatedMetagenomicData, prerequisite steps for analysis, estimation of alpha diversity, analysis of beta diversity, and differential abundance analysis. To make the tables and figures in this tutorial, a number of R packages are required, as follows.
library(curatedMetagenomicData)
library(dplyr)
library(stringr)
library(scater)
library(snakecase)
library(forcats)
library(gtsummary)
library(mia)
library(ggplot2)
library(ggsignif)
library(hrbrthemes)
library(vegan)
library(uwot)
library(ANCOMBC)
library(tibble)
library(tidyr)
library(knitr)
library(ggrepel)With the requisite R packages loaded and a basic working knowledge of metagenomics analysis (as was covered in the workshop), you’re ready to begin. Just one last note, in most of the code chunks below messages have been suppressed; more verbose output should be expected when using the R console.
Using curatedMetagenomicData
First, let’s address what the curatedMetagenomicData R/Bioconductor package is; here’s an brief description.
The curatedMetagenomicData package provides standardized, curated human microbiome data for novel analyses. It includes gene families, marker abundance, marker presence, pathway abundance, pathway coverage, and relative abundance for samples collected from different body sites. The bacterial, fungal, and archaeal taxonomic abundances for each sample were calculated with MetaPhlAn3 and metabolic functional potential was calculated with HUMAnN3. The manually curated sample metadata and standardized metagenomic data are available as (Tree)SummarizedExperiment objects.
Be sure to refer to the SummarizedExperiment and TreeSummarizedExperiment vignettes if the data structures are unclear. There is also a reference website for curatedMetagenomicData at waldronlab.io/curatedMetagenomicData, if you need it.
Sample Metadata Exploration
Any metagenomic analysis that uses curatedMetagenomicData
is likely to begin with an exploration of sample metadata
(i.e. sampleMetadata). The sampleMetadata
object is a data.frame in the package that contains sample
metadata that has been manually curated by humans for each and every
sample. To give you an idea of some of the columns that are available,
dplyr
syntax is used below to take a random sampling of 10 rows
(slice_sample). Then, columns containing NA
values are removed, and the first 10 remaining columns are selected. The
data.frame is sorted alphabetically by
study_name prior to being returned.
sampleMetadata |>
slice_sample(n = 10) |>
select(where(~ !any(is.na(.x)))) |>
select(1:10) |>
arrange(study_name)## study_name sample_id subject_id body_site
## 1 AsnicarF_2021 SAMEA7045315 predict1_MTG_0461 stool
## 2 BrooksB_2017 S2_011_011G1 S2_011 stool
## 3 HMP_2019_ibdmdb CSM67UF5 C3011 stool
## 4 HMP_2019_ibdmdb PSM6XBUO P6010 stool
## 5 LiuW_2016 SRR3992974 SRR3992974 stool
## 6 MehtaRS_2018 SID50745639_SF07 SID50745639 stool
## 7 QinN_2014 LD-98 LD-98 stool
## 8 TettAJ_2016 SK_CT108OSR_t1M14 TettAJ_2016108 skin
## 9 VatanenT_2016 G78515 T012977 stool
## 10 ZeeviD_2015 PNP_Main_772 PNP_Main_772 stool
## study_condition disease age_category country
## 1 control healthy adult GBR
## 2 premature_born premature_born newborn USA
## 3 IBD IBD adult USA
## 4 IBD IBD child USA
## 5 control healthy adult MNG
## 6 control healthy adult USA
## 7 cirrhosis ascites;cirrhosis;hepatitis adult CHN
## 8 psoriasis psoriasis adult ITA
## 9 control healthy child EST
## 10 control healthy adult ISR
## non_westernized sequencing_platform
## 1 no IlluminaNovaSeq
## 2 no IlluminaHiSeq
## 3 no IlluminaHiSeq
## 4 no IlluminaHiSeq
## 5 no IlluminaHiSeq
## 6 no IlluminaHiSeq
## 7 no IlluminaHiSeq
## 8 no IlluminaHiSeq
## 9 no IlluminaHiSeq
## 10 no IlluminaHiSeqFinding Available Resources
The resources available in curatedMetagenomicData
are organized by study_name and can be discovered with the
curatedMetagenomicData() function. When provided with a
string, it will return the names of available resources. Each
study_name will have 6 data types
(gene_families, marker_abundance,
marker_presence, pathway_abundance,
pathway_coverage, and relative_abundance),
these follow the study_name and are separated by a dot. The
date that precedes study_name is for versioning, but it’s
unimportant here.
curatedMetagenomicData("AsnicarF")## 2021-03-31.AsnicarF_2017.gene_families
## 2021-03-31.AsnicarF_2017.marker_abundance
## 2021-03-31.AsnicarF_2017.marker_presence
## 2021-03-31.AsnicarF_2017.pathway_abundance
## 2021-03-31.AsnicarF_2017.pathway_coverage
## 2021-03-31.AsnicarF_2017.relative_abundance
## 2021-10-14.AsnicarF_2017.gene_families
## 2021-10-14.AsnicarF_2017.marker_abundance
## 2021-10-14.AsnicarF_2017.marker_presence
## 2021-10-14.AsnicarF_2017.pathway_abundance
## 2021-10-14.AsnicarF_2017.pathway_coverage
## 2021-10-14.AsnicarF_2017.relative_abundance
## 2021-03-31.AsnicarF_2021.gene_families
## 2021-03-31.AsnicarF_2021.marker_abundance
## 2021-03-31.AsnicarF_2021.marker_presence
## 2021-03-31.AsnicarF_2021.pathway_abundance
## 2021-03-31.AsnicarF_2021.pathway_coverage
## 2021-03-31.AsnicarF_2021.relative_abundanceThe first argument passed to curatedMetagenomicData(),
pattern, is actually a regular expression. So things like
.+ (match any character one or more times) work, and the
names multiple resources are returned if they match. Below, you can see
there are two AsnicarF resources for the
relative_abundance data type.
curatedMetagenomicData("AsnicarF.+.relative_abundance")## 2021-03-31.AsnicarF_2017.relative_abundance
## 2021-10-14.AsnicarF_2017.relative_abundance
## 2021-03-31.AsnicarF_2021.relative_abundanceDownloading Study Resources
As is clear from the examples above, simply searching for the
AsnicarF studies did not download any curated metagenomic
data. To do that you must provide another argument,
dryrun = FALSE, to the
curatedMetagenomicData() function. Doing so will download
the matching resources from ExperimentHub
(or load them from the local cache). When dryrun = FALSE,
curatedMetagenomicData() will always return a
list of SummarizedExperiment
and/or TreeSummarizedExperiment
objects.
curatedMetagenomicData("AsnicarF.+.relative_abundance", dryrun = FALSE)## $`2021-10-14.AsnicarF_2017.relative_abundance`
## class: TreeSummarizedExperiment
## dim: 298 24
## metadata(0):
## assays(1): relative_abundance
## rownames(298):
## k__Bacteria|p__Proteobacteria|c__Gammaproteobacteria|o__Enterobacterales|f__Enterobacteriaceae|g__Escherichia|s__Escherichia_coli
## k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Bifidobacteriales|f__Bifidobacteriaceae|g__Bifidobacterium|s__Bifidobacterium_bifidum
## ...
## k__Bacteria|p__Firmicutes|c__Bacilli|o__Lactobacillales|f__Streptococcaceae|g__Streptococcus|s__Streptococcus_gordonii
## k__Bacteria|p__Firmicutes|c__Bacilli|o__Lactobacillales|f__Aerococcaceae|g__Abiotrophia|s__Abiotrophia_sp_HMSC24B09
## rowData names(7): superkingdom phylum ... genus species
## colnames(24): MV_FEI1_t1Q14 MV_FEI2_t1Q14 ... MV_MIM5_t2M14
## MV_MIM5_t3F15
## colData names(22): study_name subject_id ... pregnant lactating
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## rowLinks: a LinkDataFrame (298 rows)
## rowTree: 1 phylo tree(s) (10430 leaves)
## colLinks: NULL
## colTree: NULL
##
## $`2021-03-31.AsnicarF_2021.relative_abundance`
## class: TreeSummarizedExperiment
## dim: 639 1098
## metadata(0):
## assays(1): relative_abundance
## rownames(639):
## k__Bacteria|p__Bacteroidetes|c__Bacteroidia|o__Bacteroidales|f__Bacteroidaceae|g__Bacteroides|s__Bacteroides_vulgatus
## k__Bacteria|p__Bacteroidetes|c__Bacteroidia|o__Bacteroidales|f__Bacteroidaceae|g__Bacteroides|s__Bacteroides_stercoris
## ...
## k__Bacteria|p__Synergistetes|c__Synergistia|o__Synergistales|f__Synergistaceae|g__Pyramidobacter|s__Pyramidobacter_sp_C12_8
## k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Micrococcales|f__Brevibacteriaceae|g__Brevibacterium|s__Brevibacterium_aurantiacum
## rowData names(7): superkingdom phylum ... genus species
## colnames(1098): SAMEA7041133 SAMEA7041134 ... SAMEA7045952 SAMEA7045953
## colData names(24): study_name subject_id ... BMI family
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## rowLinks: a LinkDataFrame (639 rows)
## rowTree: 1 phylo tree(s) (10430 leaves)
## colLinks: NULL
## colTree: NULLMerging Data Across Studies
It can be useful to have the list elements returned by
curatedMetagenomicData() merged into a single SummarizedExperiment
or TreeSummarizedExperiment
object. This is accomplished using the mergeData() function
– simply pipe (|>) the output of
curatedMetagenomicData() to mergeData() and a
single object will be returned.
curatedMetagenomicData("AsnicarF.+.relative_abundance", dryrun = FALSE) |>
mergeData()## class: TreeSummarizedExperiment
## dim: 680 1122
## metadata(0):
## assays(1): relative_abundance
## rownames(680):
## k__Bacteria|p__Proteobacteria|c__Gammaproteobacteria|o__Enterobacterales|f__Enterobacteriaceae|g__Escherichia|s__Escherichia_coli
## k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Bifidobacteriales|f__Bifidobacteriaceae|g__Bifidobacterium|s__Bifidobacterium_bifidum
## ...
## k__Bacteria|p__Synergistetes|c__Synergistia|o__Synergistales|f__Synergistaceae|g__Pyramidobacter|s__Pyramidobacter_sp_C12_8
## k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Micrococcales|f__Brevibacteriaceae|g__Brevibacterium|s__Brevibacterium_aurantiacum
## rowData names(7): superkingdom phylum ... genus species
## colnames(1122): MV_FEI1_t1Q14 MV_FEI2_t1Q14 ... SAMEA7045952
## SAMEA7045953
## colData names(26): study_name subject_id ... BMI family
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## rowLinks: a LinkDataFrame (680 rows)
## rowTree: 1 phylo tree(s) (10430 leaves)
## colLinks: NULL
## colTree: NULLReturning Data from Samples
There is just one more function in the curatedMetagenomicData
package to know about, returnSamples(). It is used when you
want to return samples across studies that match a certain set of
conditions. To return a specific set of samples, you first subset the
sampleMetadata data.frame to include only the
desired samples. In the example below, stool
(body_site == "stool") samples from healthy
(disease == "healthy") adults (age >= 18)
living in Ireland or Italy (str_detect(country, "IRL|ITA"))
are included before columns of all NA values are dropped.
When the sampleMetadata data.frame is subset
as desired, it is piped to the returnSamples() function. As
you can see below, there is another argument, counts, which
has not yet been mentioned. When counts = TRUE, relative
abundance proportions are multiplied by read depth and rounded to the
nearest integer prior to being returned – the argument applies to both
returnSamples() and curatedMetagenomicData()
when requesting the relative_abundance data type.
countryData <-
filter(sampleMetadata, body_site == "stool") |>
filter(disease == "healthy") |>
filter(age >= 18) |>
filter(str_detect(country, "IRL|ITA")) |>
select(where(~ !all(is.na(.x)))) |>
returnSamples("relative_abundance", counts = TRUE)Getting Ready for Analysis
First, have a look at the countryData object you just
created to get a better understanding of what it contains. You will be
using it for the rest of this tutorial.
countryData## class: TreeSummarizedExperiment
## dim: 906 281
## metadata(0):
## assays(1): relative_abundance
## rownames(906):
## k__Bacteria|p__Bacteroidetes|c__Bacteroidia|o__Bacteroidales|f__Prevotellaceae|g__Prevotella|s__Prevotella_copri
## k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Bifidobacteriales|f__Bifidobacteriaceae|g__Bifidobacterium|s__Bifidobacterium_adolescentis
## ...
## k__Bacteria|p__Firmicutes|c__Clostridia|o__Clostridiales|f__Clostridiaceae|g__Clostridium|s__Clostridium_sp_D5
## k__Bacteria|p__Firmicutes|c__Bacilli|o__Lactobacillales|f__Lactobacillaceae|g__Lactobacillus|s__Lactobacillus_algidus
## rowData names(7): superkingdom phylum ... genus species
## colnames(281): SID01.BA.VG.2 SID01.BA.V_metag ... CRC_MR_SBJ83H_17
## CRC_MR_SBJ84H_17
## colData names(35): study_name subject_id ... dyastolic_p systolic_p
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## rowLinks: a LinkDataFrame (906 rows)
## rowTree: 1 phylo tree(s) (10430 leaves)
## colLinks: NULL
## colTree: NULLAs can be seen above, the first assay of the TreeSummarizedExperiment
is named relative_abundance. This is usually of little
consequence, but you will need to rename the assay to
counts for the transformation in the next step; do so by
setting the assayNames as follows.
assayNames(countryData) <-
"counts"Next, use logNormCounts() from the scater
package to create a second assay logcounts that contains
log-normalized species abundances.
countryData <-
logNormCounts(countryData)By taking a look again, you can see there are now two
assays as was desired.
countryData## class: TreeSummarizedExperiment
## dim: 906 281
## metadata(0):
## assays(2): counts logcounts
## rownames(906):
## k__Bacteria|p__Bacteroidetes|c__Bacteroidia|o__Bacteroidales|f__Prevotellaceae|g__Prevotella|s__Prevotella_copri
## k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Bifidobacteriales|f__Bifidobacteriaceae|g__Bifidobacterium|s__Bifidobacterium_adolescentis
## ...
## k__Bacteria|p__Firmicutes|c__Clostridia|o__Clostridiales|f__Clostridiaceae|g__Clostridium|s__Clostridium_sp_D5
## k__Bacteria|p__Firmicutes|c__Bacilli|o__Lactobacillales|f__Lactobacillaceae|g__Lactobacillus|s__Lactobacillus_algidus
## rowData names(7): superkingdom phylum ... genus species
## colnames(281): SID01.BA.VG.2 SID01.BA.V_metag ... CRC_MR_SBJ83H_17
## CRC_MR_SBJ84H_17
## colData names(36): study_name subject_id ... systolic_p sizeFactor
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## rowLinks: a LinkDataFrame (906 rows)
## rowTree: 1 phylo tree(s) (10430 leaves)
## colLinks: NULL
## colTree: NULLBefore diving into analysis, you should create a quick summary table to understand the metadata better and be prepared to handle potential biases in analysis. The steps below use dplyr syntax to clean up a few of the variables before they are output in the summary table.
colData(countryData) |>
as.data.frame() |>
select(study_name, age, gender, country, sequencing_platform, curator, diet) |>
mutate(gender = to_title_case(gender)) |>
mutate(gender = fct_explicit_na(gender)) |>
mutate(curator = str_replace_all(curator, "_", " ")) |>
mutate(curator = str_replace_all(curator, ";", ", ")) |>
mutate(diet = to_title_case(diet)) |>
mutate(diet = fct_explicit_na(diet)) |>
rename_with(to_title_case) |>
tbl_summary()## Warning: There was 1 warning in `mutate()`.
## ℹ In argument: `gender = fct_explicit_na(gender)`.
## Caused by warning:
## ! `fct_explicit_na()` was deprecated in forcats 1.0.0.
## ℹ Please use `fct_na_value_to_level()` instead.| Characteristic | N = 2811 |
|---|---|
| Study Name | |
| DeFilippisF_2019 | 93 (33%) |
| FerrettiP_2018 | 19 (6.8%) |
| KeohaneDM_2020 | 117 (42%) |
| RampelliS_2015 | 11 (3.9%) |
| ThomasAM_2018a | 14 (5.0%) |
| ThomasAM_2018b | 27 (9.6%) |
| Age | 39 (31, 52) |
| Gender | |
| Female | 157 (56%) |
| Male | 123 (44%) |
| (Missing) | 1 (0.4%) |
| Country | |
| IRL | 117 (42%) |
| ITA | 164 (58%) |
| Sequencing Platform | |
| IlluminaHiSeq | 188 (67%) |
| IlluminaNextSeq | 93 (33%) |
| Curator | |
| Francesca DeFilippis, Edoardo Pasolli | 93 (33%) |
| Paolo Manghi | 155 (55%) |
| Paolo Manghi, Marisa Metzger | 14 (5.0%) |
| Paolo Manghi, Pamela Ferretti | 19 (6.8%) |
| Diet | |
| Omnivore | 21 (7.5%) |
| Vegan | 36 (13%) |
| Vegetarian | 36 (13%) |
| (Missing) | 188 (67%) |
| 1 n (%); Median (Q1, Q3) | |
Estimating Alpha Diversity
Alpha diversity estimation seeks to quantify variation within a
sample; the Shannon index (H’) is probably the most widely used measure.
It accounts for both richness (i.e. how many types of bacteria are
present) and evenness (i.e. how equal the proportions of types is); it
is assessed below using the addAlpha() function from the
mia
package. Then, the plotColData() function from the scater
package is used to generate a basic ggplot2 plot,
which is stylized further using the ggsignif and
hrbrthemes
packages.
countryData |>
addAlpha(abund_values = "logcounts", index = "shannon") |>
plotColData(x = "country", y = "shannon", colour_by = "country", shape_by = "country") +
geom_signif(comparisons = list(c("IRL", "ITA")), test = "t.test", map_signif_level = TRUE) +
labs(
title = "Alpha Diversity by Country, Shannon Index (H')",
subtitle = "Stool Samples of Healthy Adults",
x = "Country",
y = "Alpha Diversity (H')"
) +
guides(color = guide_none(), shape = guide_none()) +
theme_minimal()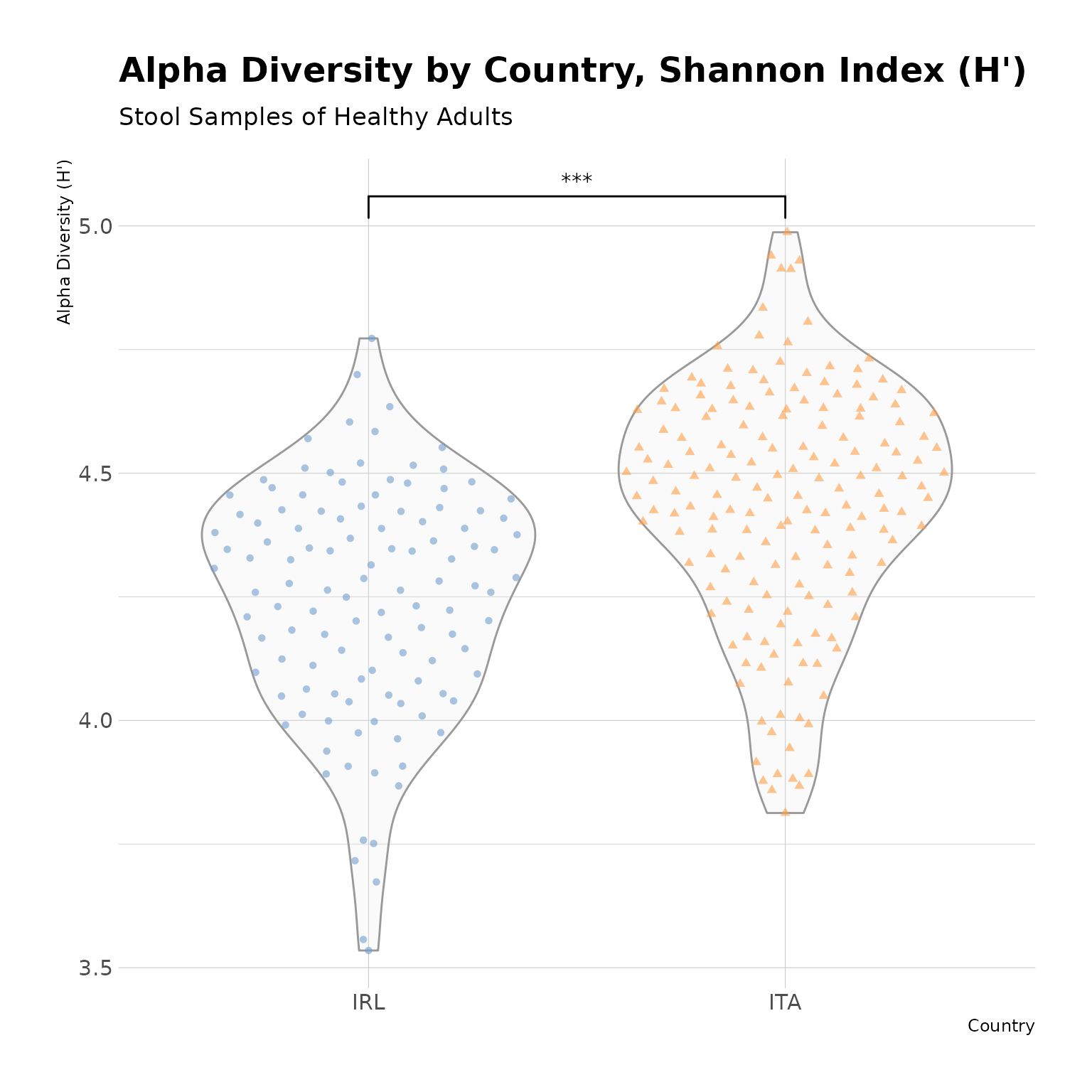
Alpha Diversity by Country. Alpha Diversity of stool samples from healthy adults, as measured by the Shannon index (H’), is significantly lower among Irish samples, as compared to Italian samples.
Analysis of Beta Diversity
Beta diversity is a measurement of between sample variation that is
usually qualitatively assessed using a low-dimensional embedding;
classically this has been done in metagenomics with a principal
coordinates analysis (PCoA) of Bray-Curtis dissimilarity.1 This is done below
using the runMDS() function from the scater
package with the vegdist function from the vegan package.
The plotReducedDim() function from the scater
package is used to generate a basic ggplot2 plot,
which is styled further.
countryData |>
runMDS(FUN = vegdist, method = "bray", exprs_values = "logcounts", name = "BrayCurtis") |>
plotReducedDim("BrayCurtis", colour_by = "country", shape_by = "country", text_by = "country") +
labs(
title = "Beta Diversity by Country, Bray-Curtis PCoA",
subtitle = "Stool Samples of Healthy Adults",
x = "PCo 1",
y = "PCo 2"
) +
guides(color = guide_none(), shape = guide_none()) +
theme_minimal()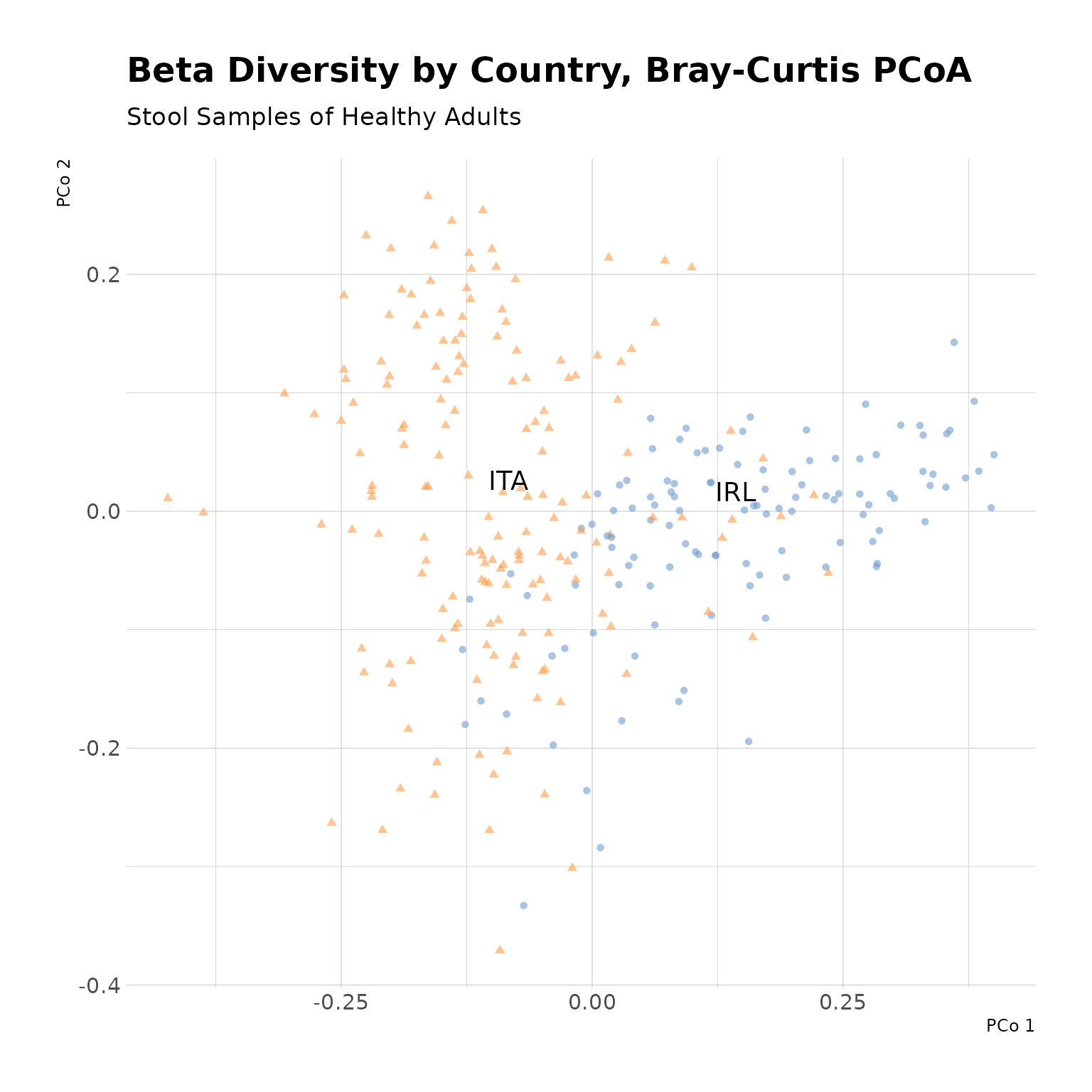
Beta Diversity by Country, Bray-Curtis PCoA. The first two principal coordinates demonstrate good seperation of Irish and Italian stool samples from healthy adults, suggesting differences in gut microbial composition between the two populations.
To address the issue of using principal coordinates analysis (PCoA)
with a dissimilarity, beta diversity can be assessed using the UMAP
(Uniform Manifold Approximation and Projection) algorithm instead.2 The
runUMAP() function from the scater
package is used below; otherwise, the syntax is largely the same as
above.
countryData |>
runUMAP(exprs_values = "logcounts", name = "UMAP") |>
plotReducedDim("UMAP", colour_by = "country", shape_by = "country", text_by = "country") +
labs(
title = "Beta Diversity by Country, UMAP Embedding",
subtitle = "Stool Samples of Healthy Adults",
x = "UMAP 1",
y = "UMAP 2"
) +
guides(color = guide_none(), shape = guide_none()) +
theme_minimal()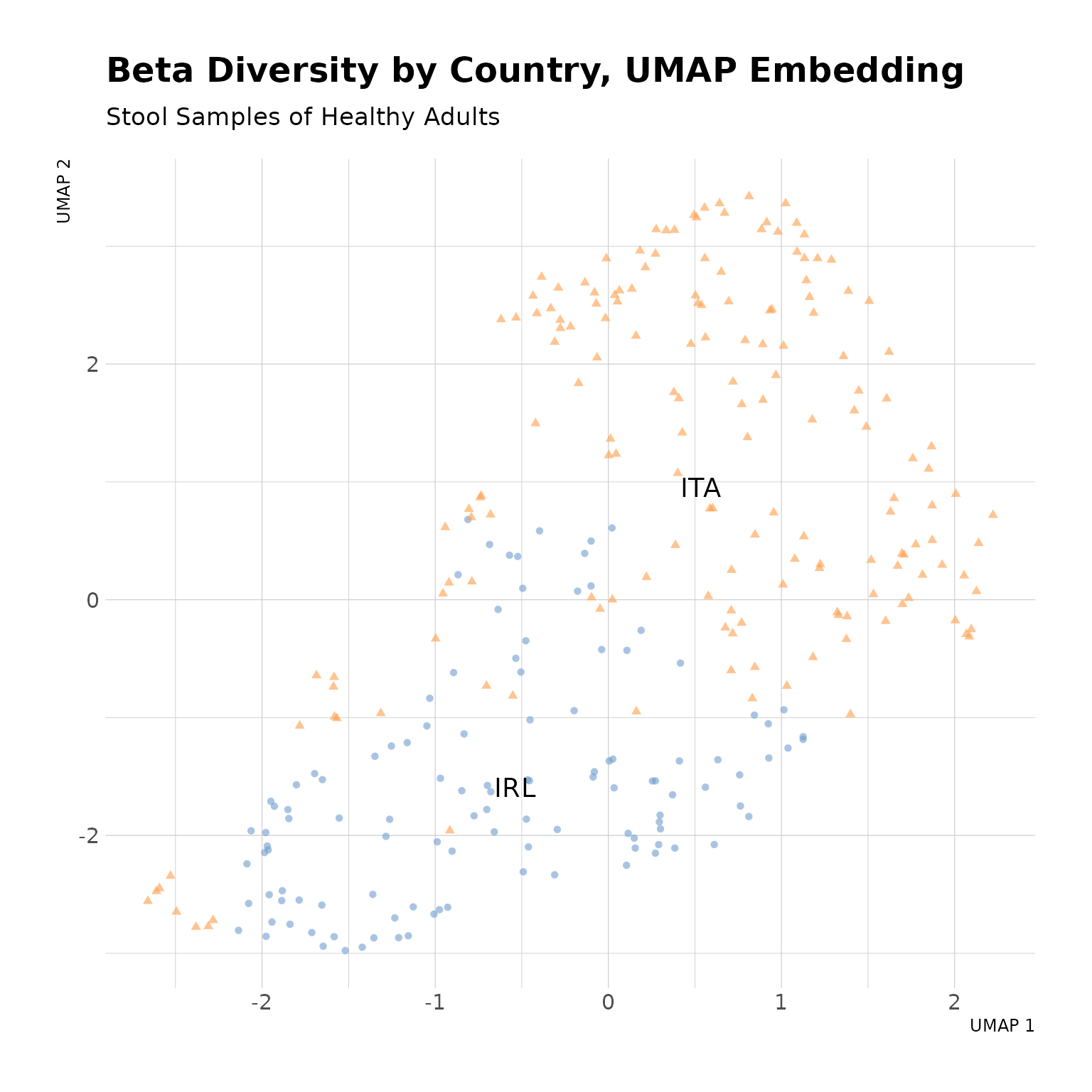
Beta Diversity by Country, UMAP Embedding. The two-dimensional UMAP embedding demonstrates good seperation of Irish and Italian stool samples from healthy adults, suggesting differences in gut microbial composition between the two populations.
Modeling Bacteria by Country
Where beta diversity embeddings suggest there are features that
separate Irish and Italian stool samples from healthy adults, you might
like to know which are most differentially abundance. To assess this the
ANCOMBC
package has a metagenomics-specific additive log-ratio model for the
task. However, the ancombc() function requires a
phyloseq class objects – one can be created using the
makePhyloseqFromTreeSummarizedExperiment() function from
the mia package,
as shown below.
ancombcResults <-
mia::convertToPhyloseq(countryData) |>
ancombc(formula="country", group="country")The results of the ANCOMBC
model are in a strange list structure and have to be
coerced into a data.frame before they can be displayed; the
bind_cols() function from the dplyr package
is used below.
# Extract the results for the country coefficient (countryITA)
ancombcTable <- data.frame(
beta = ancombcResults$res$lfc$countryITA,
se = ancombcResults$res$se$countryITA,
p_val = ancombcResults$res$p_val$countryITA,
q_val = ancombcResults$res$q_val$countryITA,
row.names = rownames(ancombcResults$feature_table)
)The row names of the results table are big long strings of microbial
taxonomies that will need some editing if they are to be displayed
nicely. the rownames_to_column() function from the tibble package
is used below to turn them into a column so they can be edited.
ancombcTable <- rownames_to_column(ancombcTable)Before the row names are split into 7 pieces, the names of columns that each piece will be assigned to are created below.
rankNames <-
c("Kingdom", "Phylum", "Class", "Order", "Family", "Genus", "Species")The row names of the results table are then transformed using tidyr, dplyr, and stringr, as shown below.
ancombcTable[["rowname"]] <-
separate(ancombcTable, rowname, rankNames, sep = "\\|") |>
select(all_of(rankNames)) |>
mutate(across(everything(), ~ str_remove_all(.x, ".__"))) |>
mutate(across(everything(), ~ str_replace_all(.x, "_", " "))) |>
mutate(label = Species) |>
pull(label)Once the results table is in good shape, it can be filtered to
include only bacterial species that exhibited large
(e.g. abs(beta) > 1) and significant
(-log10(q_val) > 5) differences in abundances between
the two countries. In the example below, the table is sorted by effect
size and a number of formatting conventions are applied before
displaying the results table.
filter(ancombcTable, abs(beta) > 1) |>
filter(-log10(q_val) > 5) |>
select(rowname, beta, se, p_val, q_val) |>
arrange(-abs(beta)) |>
column_to_rownames() |>
mutate(across(everything(), .fns = ~ round(.x, digits = 3))) |>
mutate(beta = format(beta)) |>
mutate(beta = str_replace(beta, " ", " ")) |>
mutate(p_val = if_else(p_val == 0, "< 0.001", format(p_val, nsmall = 3))) |>
mutate(q_val = if_else(q_val == 0, "< 0.001", format(q_val, nsmall = 3))) |>
kable(col.names = c("β", "SE", "P", "Q"), align = "cccc", escape = FALSE)| β | SE | P | Q | |
|---|---|---|---|---|
| Catenibacterium mitsuokai | -8.872 | 0.615 | < 0.001 | < 0.001 |
| Holdemanella biformis | -8.159 | 0.574 | < 0.001 | < 0.001 |
| Eubacterium sp CAG 180 | -7.331 | 0.694 | < 0.001 | < 0.001 |
| Bacteroides dorei | 6.269 | 0.558 | < 0.001 | < 0.001 |
| Lactobacillus ruminis | -6.088 | 0.562 | < 0.001 | < 0.001 |
| Phascolarctobacterium succinatutens | -5.970 | 0.712 | < 0.001 | < 0.001 |
| Ruminococcus torques | -5.703 | 0.450 | < 0.001 | < 0.001 |
| Alistipes putredinis | 5.690 | 0.624 | < 0.001 | < 0.001 |
| Eubacterium hallii | -5.384 | 0.444 | < 0.001 | < 0.001 |
| Bifidobacterium angulatum | -5.301 | 0.549 | < 0.001 | < 0.001 |
| Prevotella sp CAG 279 | -5.122 | 0.630 | < 0.001 | < 0.001 |
| Dorea formicigenerans | -4.970 | 0.374 | < 0.001 | < 0.001 |
| Lawsonibacter asaccharolyticus | 4.946 | 0.413 | < 0.001 | < 0.001 |
| Methanobrevibacter smithii | -4.735 | 0.640 | < 0.001 | < 0.001 |
| Prevotella sp 885 | -4.697 | 0.590 | < 0.001 | < 0.001 |
| Coprococcus comes | -4.589 | 0.424 | < 0.001 | < 0.001 |
| Oscillibacter sp CAG 241 | -4.478 | 0.546 | < 0.001 | < 0.001 |
| Roseburia intestinalis | -4.462 | 0.566 | < 0.001 | < 0.001 |
| Slackia isoflavoniconvertens | -4.422 | 0.586 | < 0.001 | < 0.001 |
| Eubacterium ramulus | -4.376 | 0.495 | < 0.001 | < 0.001 |
| Ruminococcus callidus | -4.366 | 0.519 | < 0.001 | < 0.001 |
| Coprococcus catus | -4.316 | 0.429 | < 0.001 | < 0.001 |
| Intestinibacter bartlettii | -4.227 | 0.536 | < 0.001 | < 0.001 |
| Eubacterium sp CAG 251 | -3.925 | 0.657 | < 0.001 | < 0.001 |
| Dorea longicatena | -3.896 | 0.332 | < 0.001 | < 0.001 |
| Alistipes finegoldii | 3.819 | 0.547 | < 0.001 | < 0.001 |
| Prevotella sp AM42 24 | -3.603 | 0.563 | < 0.001 | < 0.001 |
| Roseburia faecis | -3.573 | 0.497 | < 0.001 | < 0.001 |
| Firmicutes bacterium CAG 170 | -3.390 | 0.582 | < 0.001 | < 0.001 |
| Bacteroides plebeius | 3.291 | 0.529 | < 0.001 | < 0.001 |
| Alistipes shahii | 3.248 | 0.535 | < 0.001 | < 0.001 |
| Bacteroides eggerthii | 3.135 | 0.483 | < 0.001 | < 0.001 |
| Parasutterella excrementihominis | 3.117 | 0.485 | < 0.001 | < 0.001 |
| Blautia wexlerae | -3.111 | 0.434 | < 0.001 | < 0.001 |
| Eubacterium rectale | -3.084 | 0.413 | < 0.001 | < 0.001 |
| Parabacteroides distasonis | 3.069 | 0.530 | < 0.001 | < 0.001 |
| Clostridium disporicum | -3.037 | 0.443 | < 0.001 | < 0.001 |
| Butyricimonas virosa | 3.035 | 0.462 | < 0.001 | < 0.001 |
| Alistipes indistinctus | 2.945 | 0.522 | < 0.001 | < 0.001 |
| Anaerostipes hadrus | -2.895 | 0.463 | < 0.001 | < 0.001 |
| Bacteroides sp CAG 530 | -2.847 | 0.413 | < 0.001 | < 0.001 |
| Blautia obeum | -2.833 | 0.410 | < 0.001 | < 0.001 |
| Intestinimonas butyriciproducens | 2.698 | 0.469 | < 0.001 | < 0.001 |
| Roseburia inulinivorans | -2.586 | 0.369 | < 0.001 | < 0.001 |
| Prevotella stercorea | -2.571 | 0.460 | < 0.001 | < 0.001 |
| Collinsella aerofaciens | -2.364 | 0.323 | < 0.001 | < 0.001 |
| Fusicatenibacter saccharivorans | -1.571 | 0.274 | < 0.001 | < 0.001 |
While the table above is somewhat interesting, the results are better summarized as a volcano plot (i.e. statistical significance versus fold change) and one can be made using the results table. To shorten labels for readability, stringr is first used to abbreviate species names by replacing the first word of the name with a single letter followed by a period. Next, the same filtering of the table as above is undertaken, and the color scheme used in all the plots above is applied. Both labeling and coloring are only applied where effect size and significance thresholds are met, as denoted by the dotted lines.
ancombcTable |>
mutate(rowname = str_replace(rowname, "^([A-Z])([a-z])+ ", "\\1. ")) |>
mutate(q_val = -log10(q_val)) |>
mutate(label = if_else(abs(beta) > 1, rowname, NA_character_)) |>
mutate(label = if_else(q_val > 5, label, NA_character_)) |>
mutate(color = if_else(beta > 0, "#FF9E4A", "#729ECE")) |>
mutate(color = if_else(is.na(label), "#000000", color)) |>
ggplot(mapping = aes(beta, q_val, color = I(color), label = label, shape = I(1))) +
geom_point() +
geom_hline(linetype = "dotted", yintercept = 5) +
geom_vline(linetype = "dotted", xintercept = 1) +
geom_vline(linetype = "dotted", xintercept = -1) +
geom_label_repel(min.segment.length = 0, force = 10, max.overlaps = 20, na.rm = TRUE) +
labs(
title = "Significance vs. Effect Size, ANCOM-BC",
subtitle = "Stool Samples of Healthy Adults",
x = expression(beta),
y = expression(-~log[10]~Q)
) +
guides(color = guide_none(), shape = guide_none()) +
theme_minimal()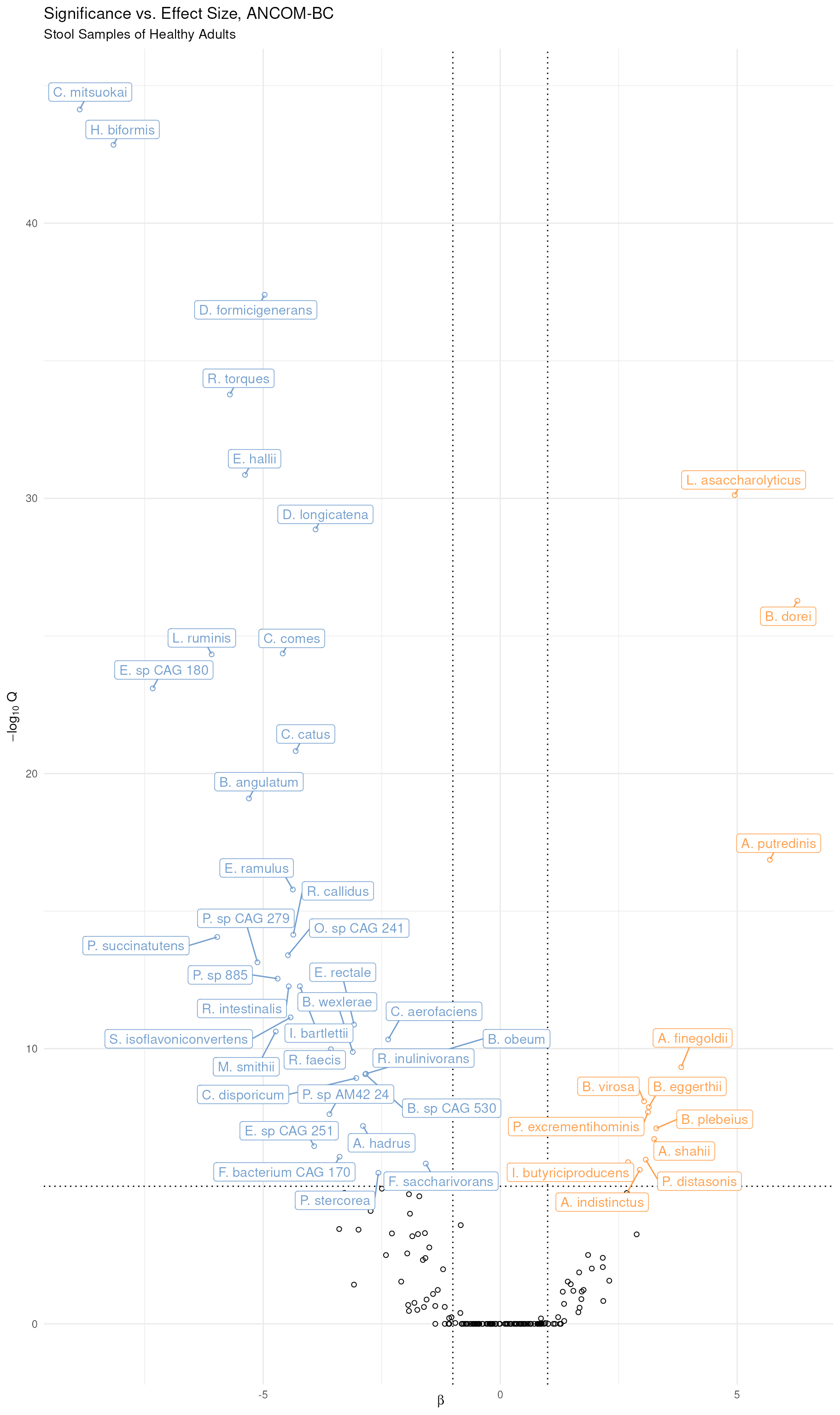
Volcano Plot of Differentially Abundance Bacterial Species. In the model and the figure, Irish samples are the reference group such that bacterial species in blue at the left are significantly more abundant in Irish samples and those in yellow at the right are significantly more abundant in Italian samples.
As a bonus, do a Google or a PubMed search for Holdemanella biformis to see what condition(s) it is associated with and then explore differences between the two countries using GBD Compare visualizations.