Applied Statistics for High-throughput Biology Session 2
Levi Waldron
day2_unsupervised.RmdDay 2 outline
Book chapter 8:
- Distances in high dimensions
- Principal Components Analysis and Singular Value Decomposition
- Multidimensional Scaling
- t-SNE and UMAP
Schematic of a typical scRNA-seq analysis workflow
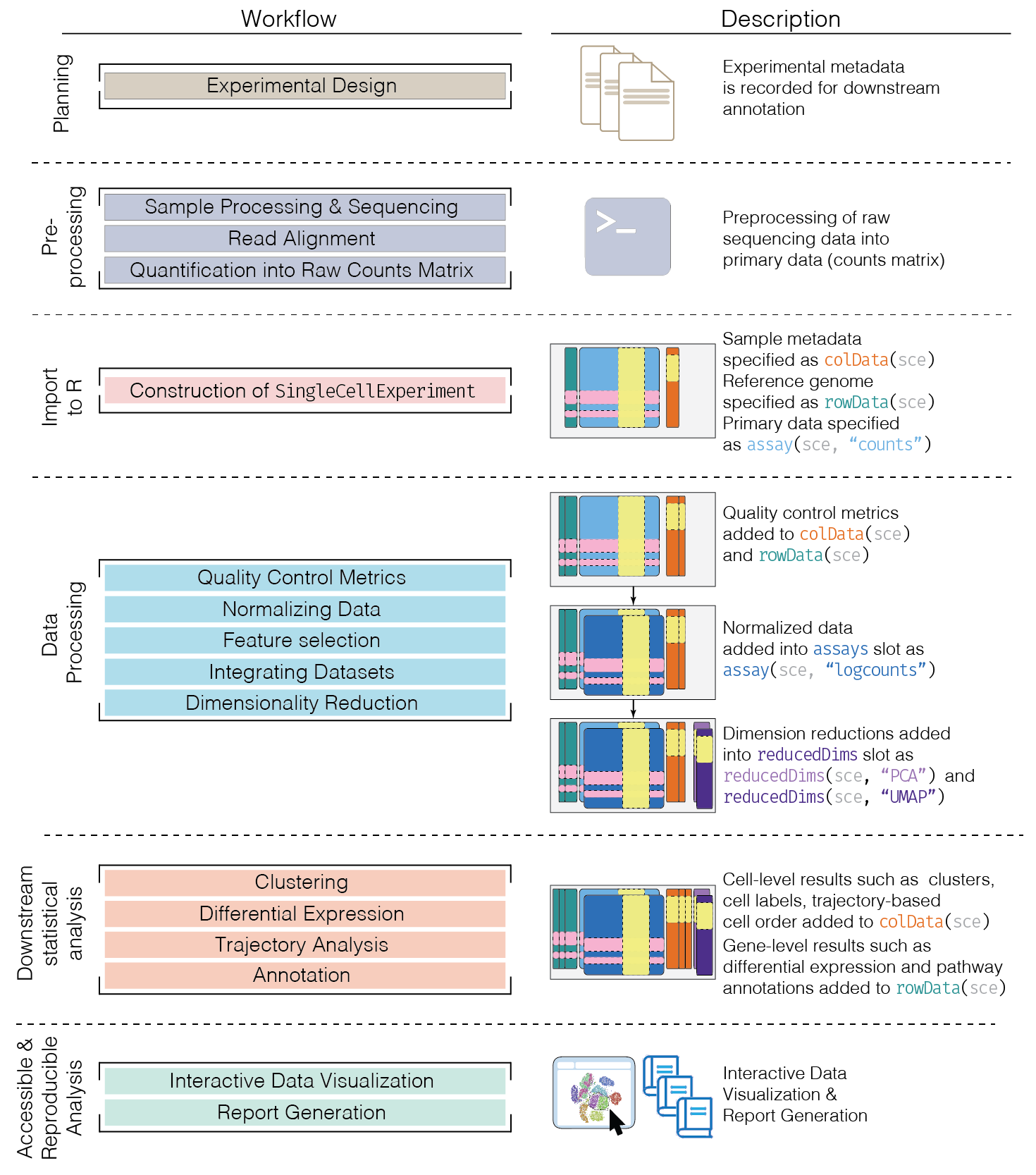
Each stage (separated by dashed lines) consists of a number of specific steps, many of which operate on and modify a SingleCellExperiment instance. (original image)
How do we measure similarity?
- Defining similarity requires choosing a metric.
- There is an infinite number of ways to do this, depending on:
- Which features to include and how they are weighted
- How features are scaled or transformed (e.g., standardizing features so they are equally important)
- The mathematical formula for the metric
Common distance metrics (Continuous data)
- Euclidean distance ( norm): Straight-line distance
- Manhattan distance ( norm): Sum of absolute differences in all coordinates
- Maximum distance ( norm): Maximum of the absolute differences across all coordinates
- Minkowski distance ( norm): Generalization of Euclidean and Manhattan
- Mahalanobis / Weighted Euclidean: Accounts for dynamic range and correlation between features
https://www.huber.embl.de/msmb/05-chap.html#how-do-we-measure-similarity
Distances for categorical, binary, and correlated data
- Edit / Hamming distance: Number of differences between character sequences
- Binary distance: Proportion of features with only one bit on
- Jaccard distance: Focuses on co-occurrence rather than co-absence (useful for ecological or mutation data)
- Correlation-based distance: Distance based on Pearson correlation
Euclidian distance (metric)
- Remember grade school:
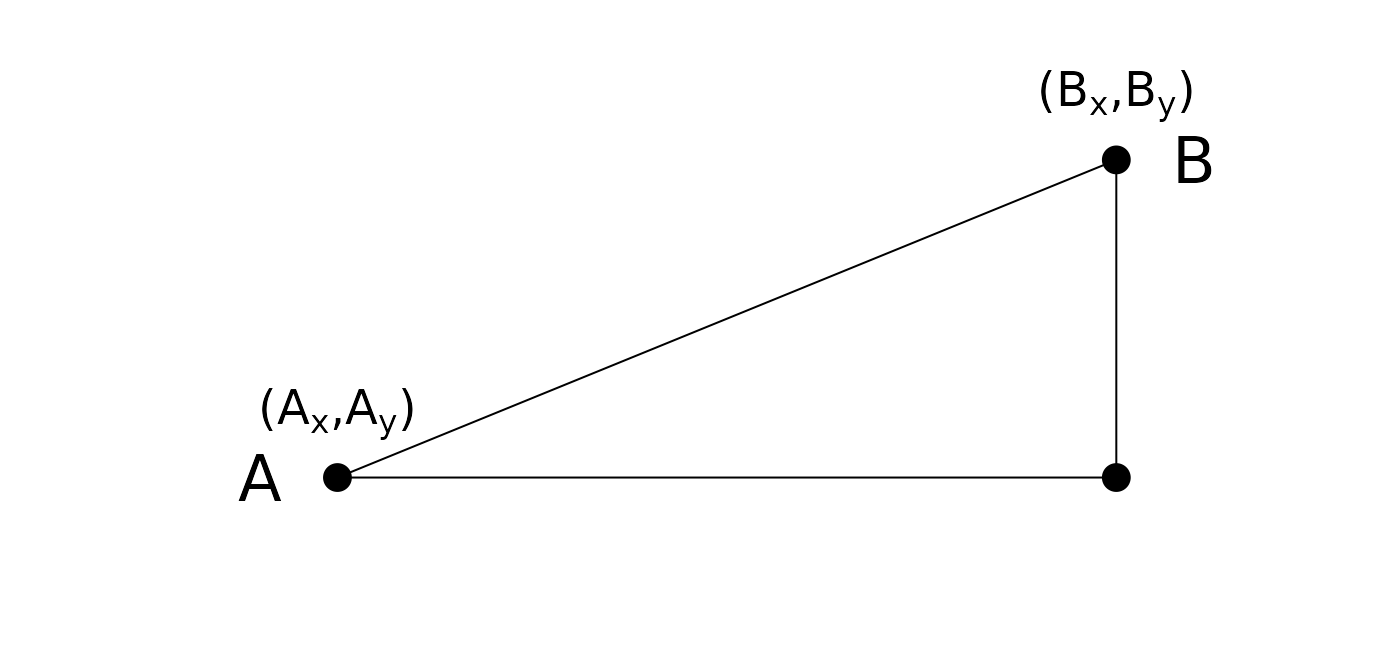
Euclidean d = . - Side note: also referred to as norm
Euclidian distance in high dimensions
This familiar concept from geometry extends directly into higher dimensions, which is essential for analyzing complex biological datasets.
## BiocManager::install("genomicsclass/tissuesGeneExpression") #if needed
## BiocManager::install("genomicsclass/GSE5859") #if needed
library(GSE5859)
library(tissuesGeneExpression)
data(tissuesGeneExpression)
# Modern approach: store expression and metadata together
sce <- SingleCellExperiment(
assays = list(logcounts = e),
colData = S4Vectors::DataFrame(tissue = tissue)
)
dim(sce) ## gene expression data
#> [1] 22215 189
table(sce$tissue) ## samples per tissue
#>
#> cerebellum colon endometrium hippocampus kidney liver
#> 38 34 15 31 39 26
#> placenta
#> 6Interested in identifying similar samples and similar genes
Notes about Euclidian distance in high dimensions
- Points are no longer on the Cartesian plane
- instead they are in higher dimensions. For example:
- sample is defined by a point in 22,215 dimensional space: .
- feature is defined by a point in 189 dimensions
Euclidean distance as for two dimensions. E.g., the distance between two samples and is:
where the sum runs over all genes, and the distance between two features and is:
where the sum runs over all samples.
Euclidian distance in matrix algebra notation
The Euclidian distance between samples and can be written as:
with and columns and .
Decomposing large matrices
- for very large matricies, consider:
- setting the
nuandnvarguments to thesvd()function - the Matrix CRAN package (sparse matrices)
- the rhdf5 and DelayedArray Bioconductor packages (on-disk arrays)
- setting the
- See note about BLAS at end
The dist() function
Excerpt from ?dist:
dist(
x,
method = "euclidean",
diag = FALSE,
upper = FALSE,
p = 2
)-
method: the distance measure to be used.
- This must be one of “euclidean”, “maximum”, “manhattan”, “canberra”, “binary” or “minkowski”. Any unambiguous substring can be given.
-
distclass output fromdist()is used for many clustering algorithms and heatmap functions
Caution: dist(e) creates a 22215 x 22215 matrix
that will probably crash your R session.
Note on standardization
- In practice, features (e.g. genes) are typically “standardized”, i.e. scaled and centered, i.e. converted to z-score:
- This is done because the differences in overall levels between
features are often not due to biological effects but technical ones,
e.g.:
- GC bias, PCR amplification efficiency, …
- Also, usually only “highly variable genes” are used to avoid scaling noise
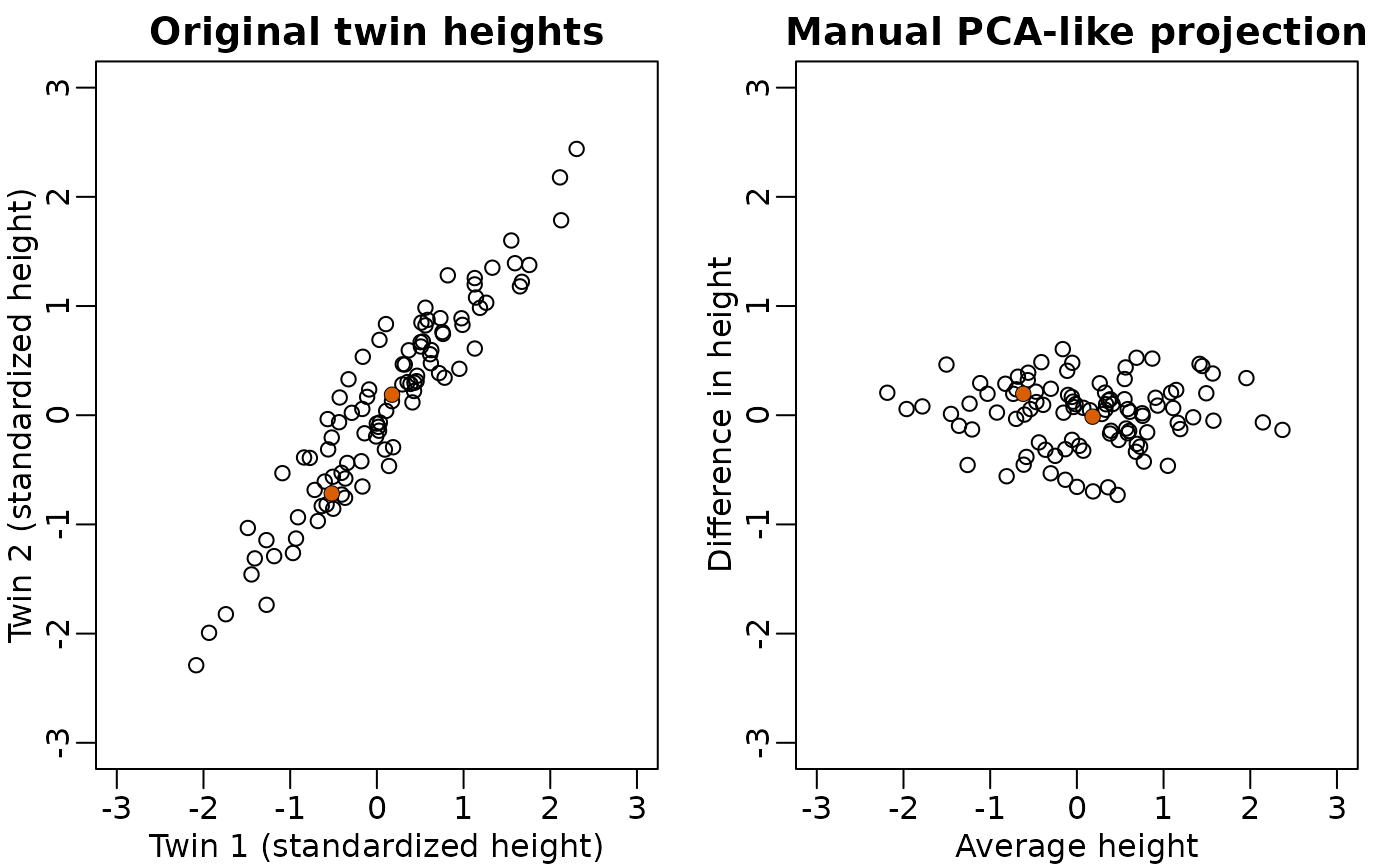
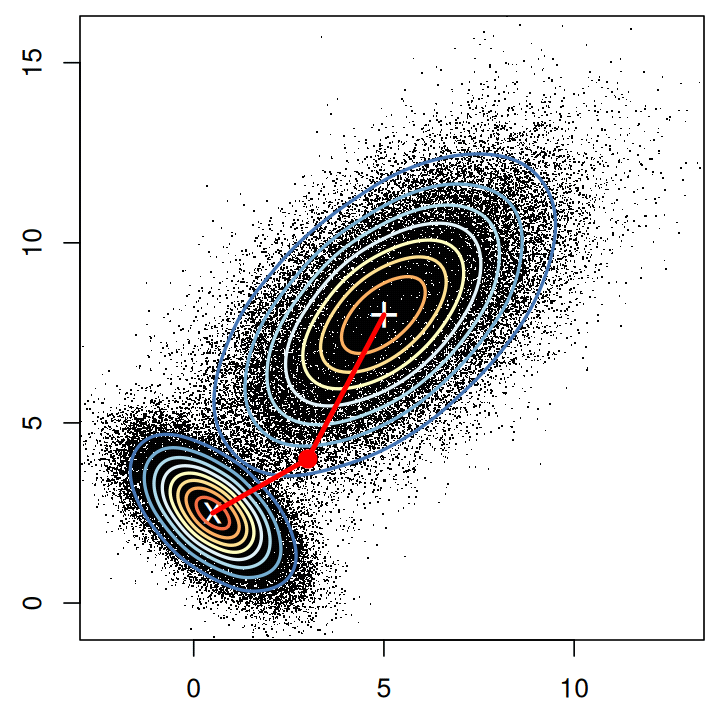
Not much distance is lost in the second dimension

- Not much loss of height differences when just using average heights
of twin pairs.
- because twin heights are highly correlated
Singular Value Decomposition (SVD)
SVD generalizes the example rotation we looked at:
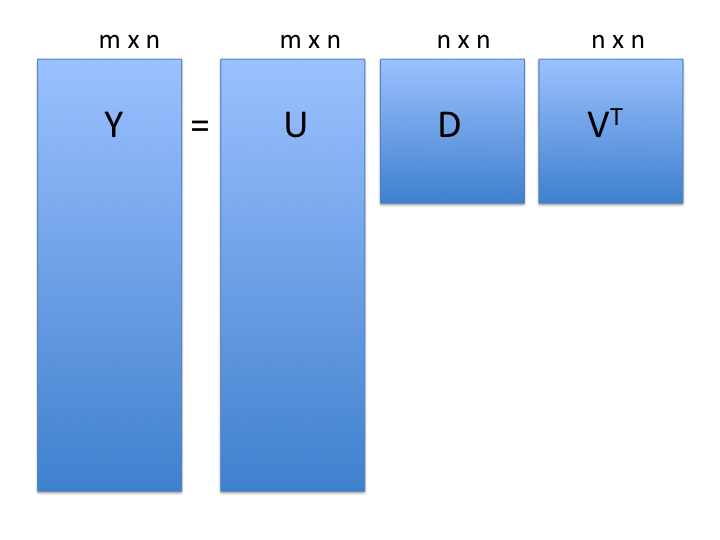
note: the above formulation is for rows columns
: the rows x cols matrix of measurements
: matrix relating original scores to PCA scores (loadings)
: diagonal matrix (eigenvalues)
-
: orthogonal matrix (eigenvectors or PCA scores)
- orthogonal = unit length and “perpendicular” in 3-D
SVD of gene expression dataset
Center but do not scale, just to make plots before more legible:
SVD:
Components of SVD results
dim(s$u) # loadings
#> [1] 22215 189
length(s$d) # eigenvalues
#> [1] 189
dim(s$v) # d %*% vT = scores
#> [1] 189 189
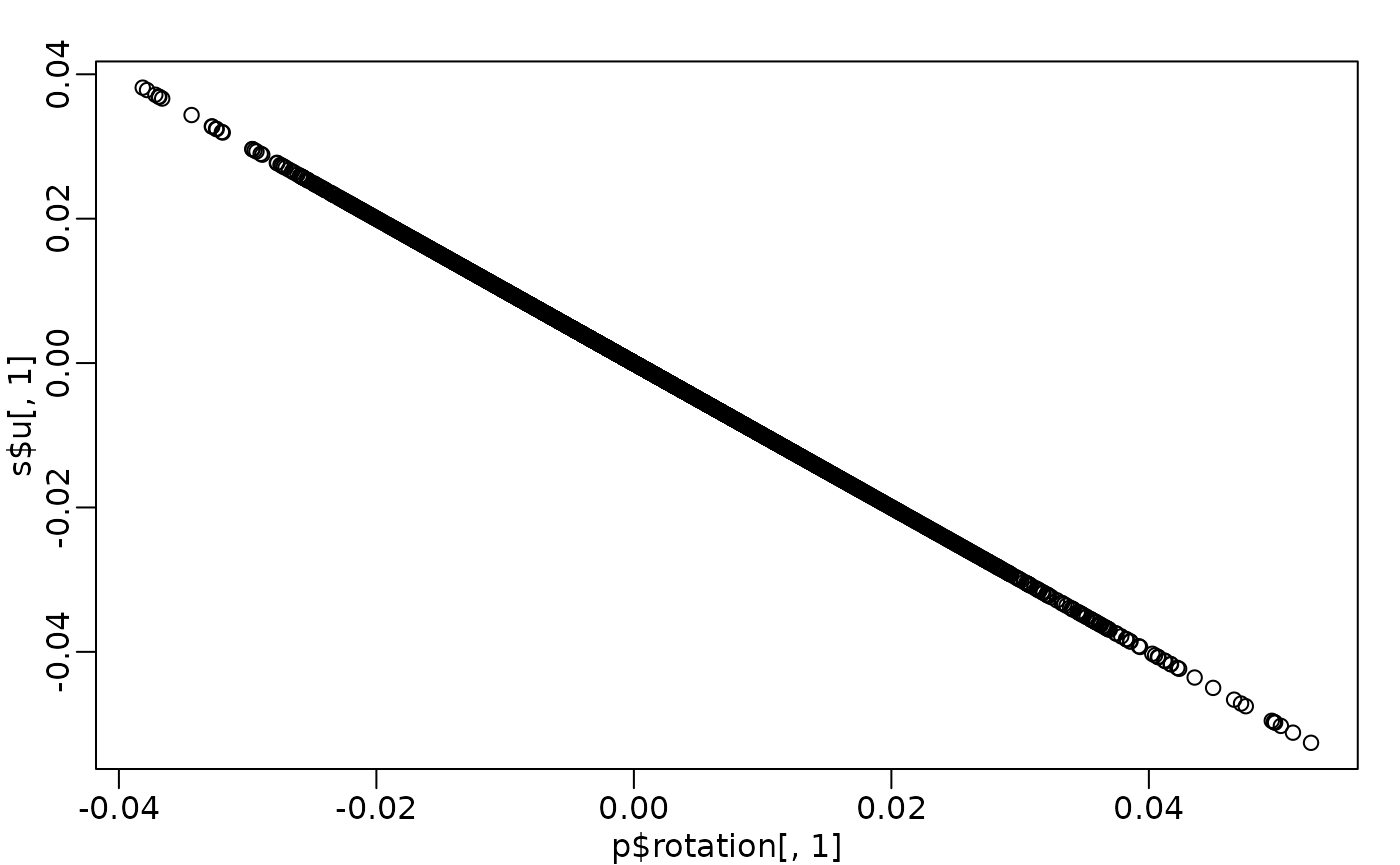
PCA interpretation: loadings
-
(loadings): relate the principal component
axes to the original variables
- think of principal component axes as a weighted combination of original axes
Visualizing PCA loadings
df_loadings <- data.frame(Index = seq_len(nrow(p$rotation)), Loading = p$rotation[, 1])
ggplot(df_loadings, aes(x = Index, y = Loading)) +
geom_point(alpha = 0.5, size = 1) +
geom_hline(yintercept = c(-0.03, 0.03), color = "red", linetype = "dashed") +
labs(title = "PC1 loadings of each gene", x = "Index of genes", y = "Loadings of PC1") +
theme_minimal()
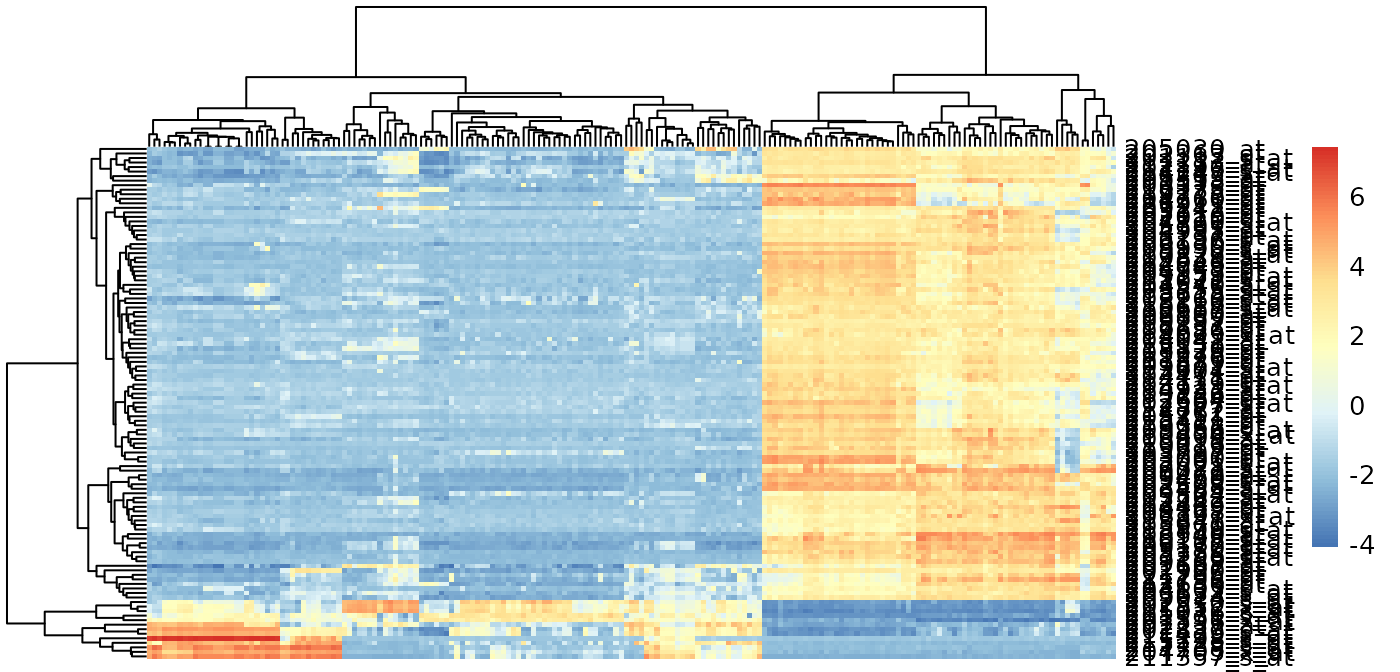
Genes with high PC1 loadings
e.pc1genes <-
e.standardize[p$rotation[, 1] < -0.03 |
p$rotation[, 1] > 0.03, ]
pheatmap::pheatmap(
e.pc1genes,
scale = "none",
show_rownames = TRUE,
show_colnames = FALSE
)
PCA interpretation: eigenvalues
- (eigenvalues): standard deviation scaling factor that each decomposed variable is multiplied by.
df_scree <- data.frame(
PC = seq_along(p$sdev),
Variance = p$sdev^2 / sum(p$sdev^2) * 100,
Cumulative = cumsum(p$sdev^2) / sum(p$sdev^2) * 100
)
ggplot(df_scree[1:150, ], aes(x = PC, y = Variance)) +
geom_line() +
geom_point(size = 1) +
labs(title = "Screeplot", y = "% variance explained") +
theme_minimal()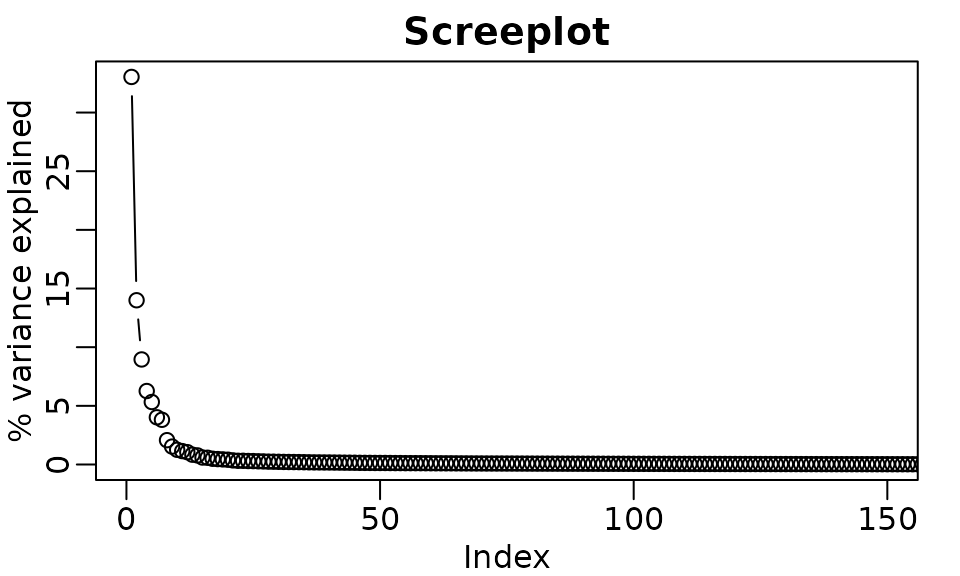
PCA interpretation: eigenvalues
Alternatively as cumulative % variance explained (using
cumsum() function)
ggplot(df_scree, aes(x = PC, y = Cumulative)) +
geom_line() +
labs(title = "Cumulative screeplot", y = "Cumulative % variance explained") +
coord_cartesian(ylim = c(0, 100)) +
theme_minimal()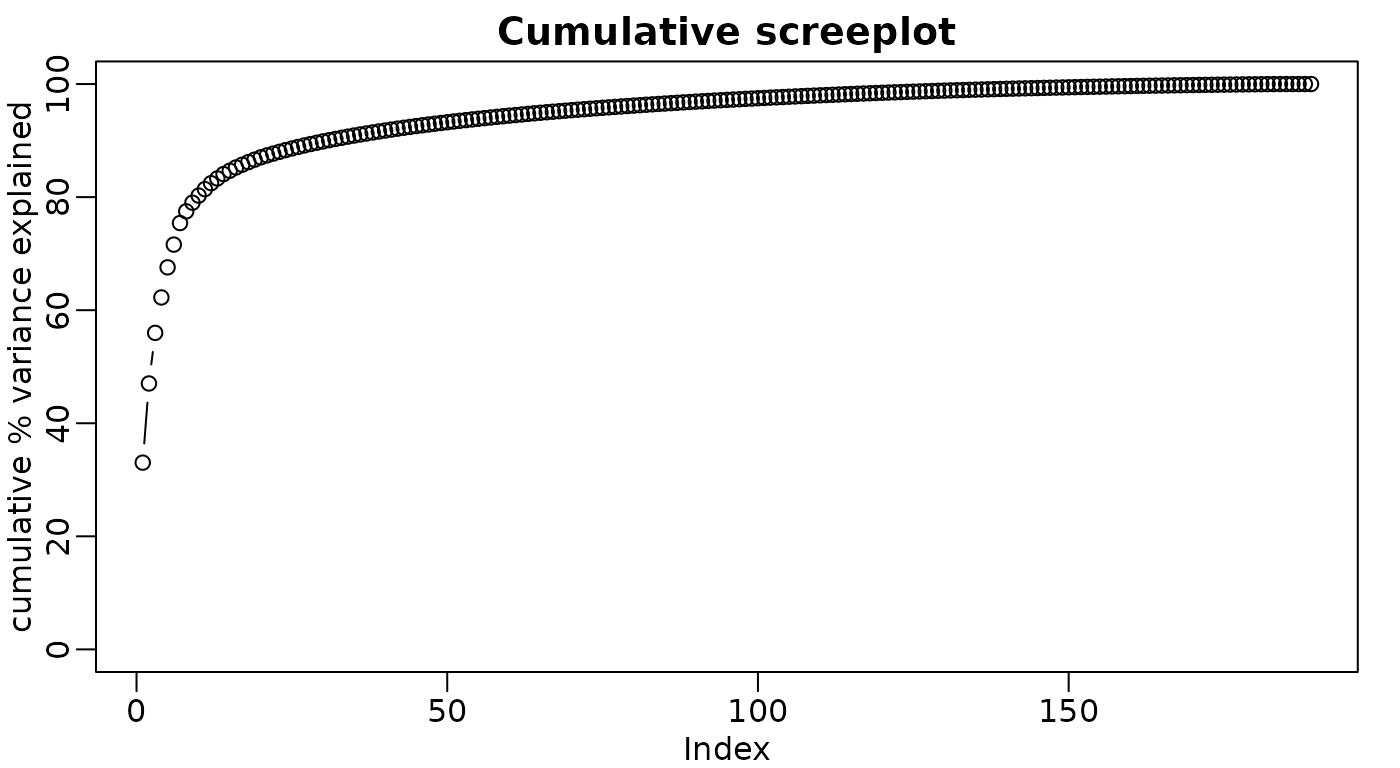
PCA interpretation: scores
- (scores): The “datapoints” in the reduced prinipal component space
- In some implementations (like
prcomp()), scores are already scaled by eigenvalues:

Multi-dimensional Scaling (MDS)
- also referred to as Principal Coordinates Analysis (PCoA)
- a reduced SVD, performed on a distance matrix
- identify two (or more) eigenvalues/vectors that preserve distances
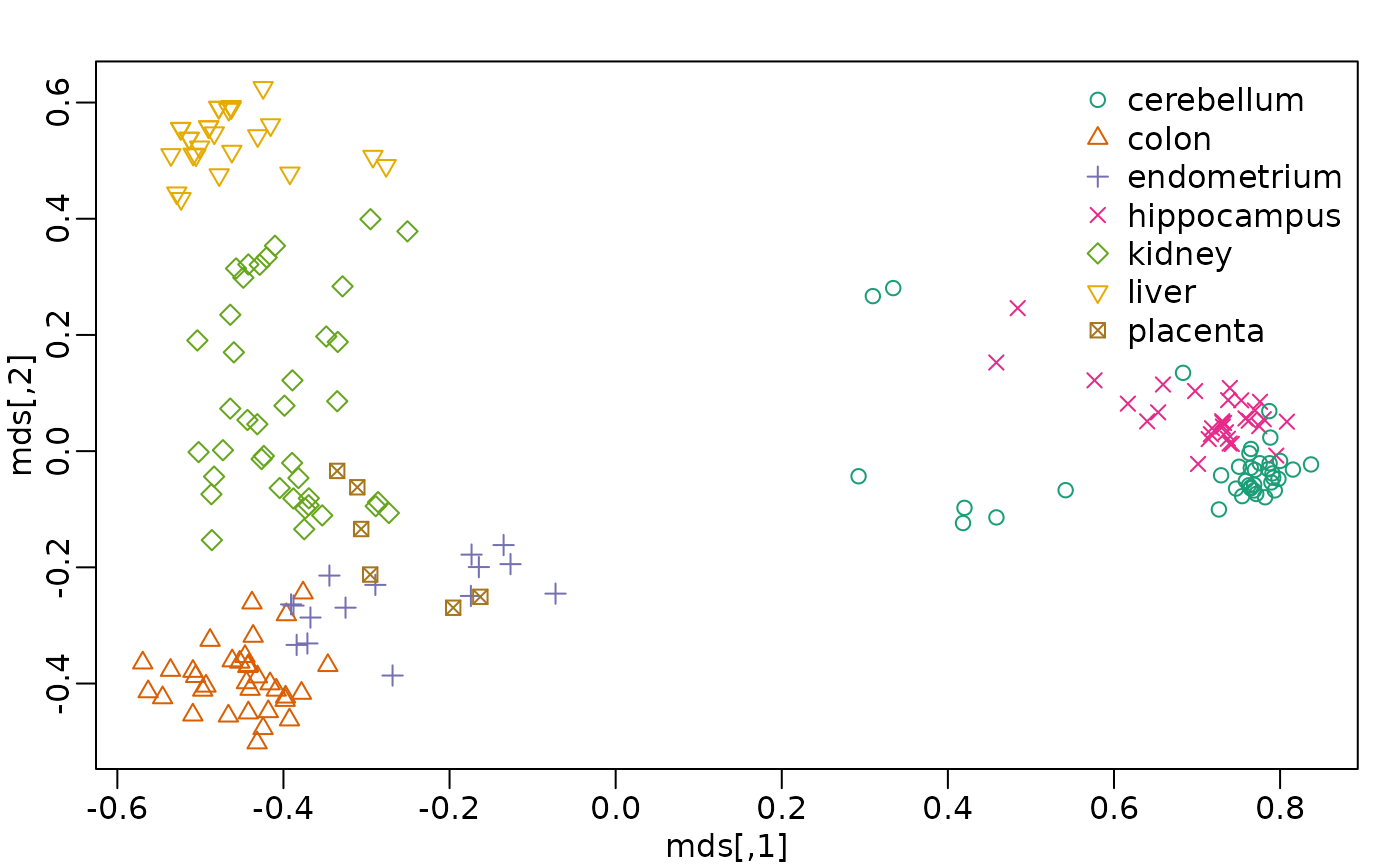
t-SNE
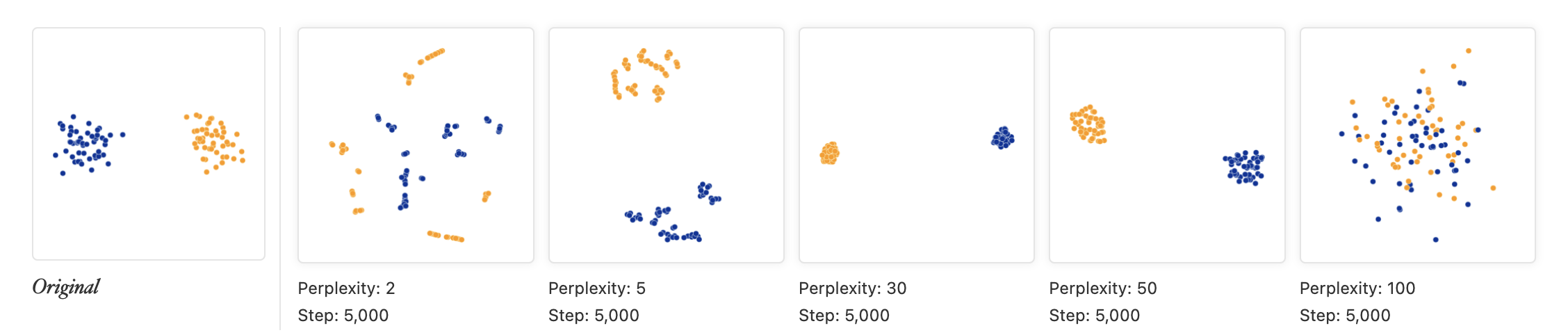
- non-linear dimension reduction method very popular for visualizing
single-cell data
- almost magical ability to show clearly separated clusters
- performs different transformations on different regions
- computationally intensive so usually done only on top ~30 PCs
- t-SNE is sensitive to choices of tuning parameters
- “perplexity” parameter defines (loosely) how to balance attention between local and global aspects of data
- optimal choice of perplexity changes for different numbers of cells from the same sample.
- perplexity = is one rule of thumb. is another (default of Rtsne)
- Here is a good post by Nikolay Oskolkov on this topic.
t-SNE caveats
- uses a random number generator
- apparent spread of clusters is completely meaningless
- distance between clusters might also not mean anything
- parameters can be tuned to make data appear how you want
- can show apparent clusters in random noise. Should not be used for statistical inference
- Try it to gain some intuition: https://distill.pub/2016/misread-tsne/)
PCA of Zeisel single-cell RNA-seq dataset
sce.zeisel <- fixedPCA(sce.zeisel, subset.row = NULL)
#> Warning in fixedPCA(sce.zeisel, subset.row = NULL): 'fixedPCA' is deprecated.
#> Use 'scrapper::runPca.se' instead.
#> See help("Deprecated")
plotReducedDim(sce.zeisel, dimred = "PCA", colour_by = "level1class")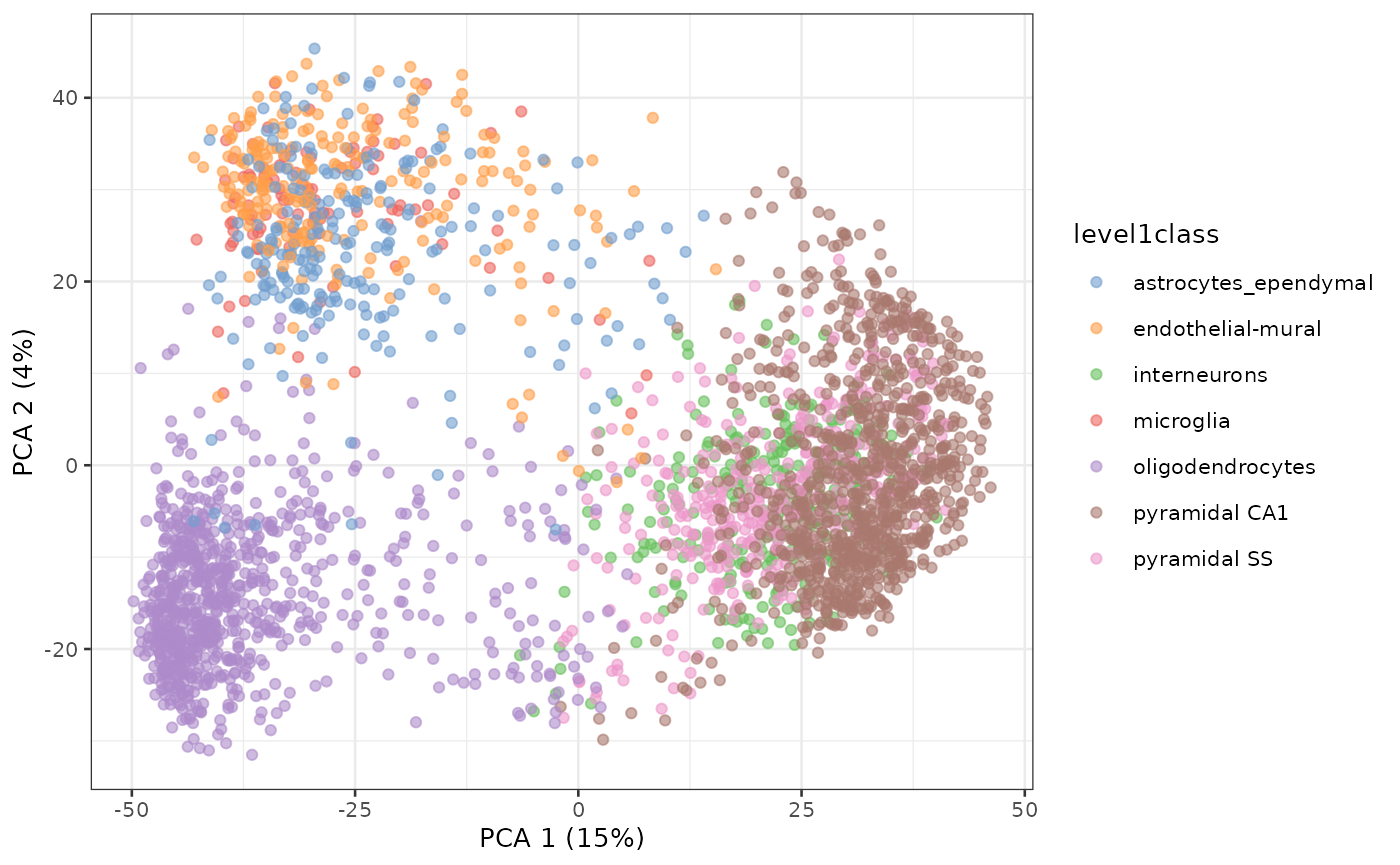
Principal Components Analysis of Zeisel dataset
t-SNE of the same dataset
sce.zeisel <- runTSNE(sce.zeisel, dimred = "PCA")
plotReducedDim(sce.zeisel, dimred = "TSNE", colour_by = "level1class")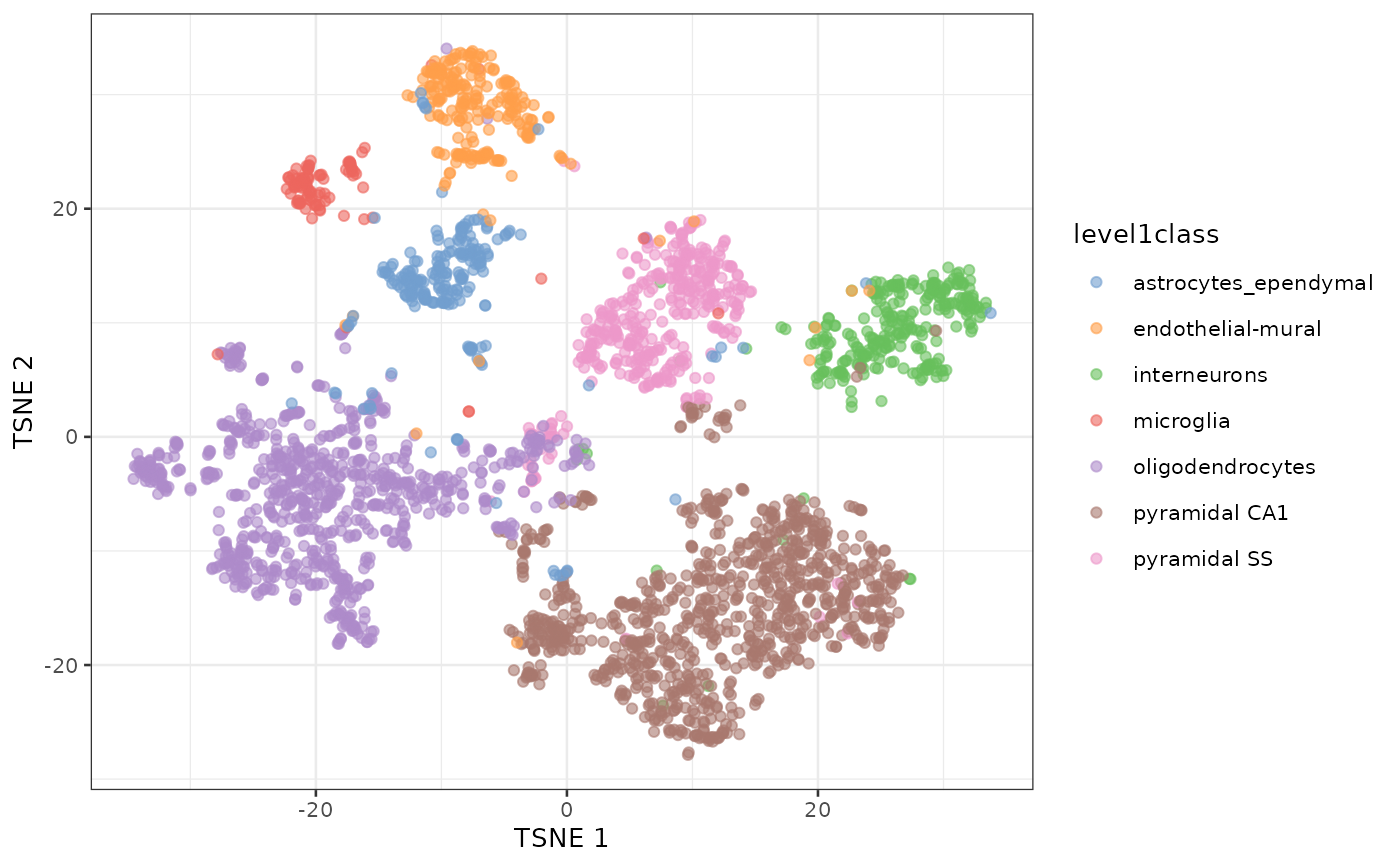
t-SNE clustering of Zeisel dataset
UMAP vs t-SNE
- UMAP may better preserve local and global distances
- tends to have more compact visual clusters with more empty space between them
- more computationally efficient
- also involves random number generation
- Note: I prefer not setting the random number seed during exploratory analysis in order to see the random variability
UMAP of the same dataset
sce.zeisel <- runUMAP(sce.zeisel, dimred = "PCA")
plotReducedDim(sce.zeisel, dimred = "UMAP", colour_by = "level1class")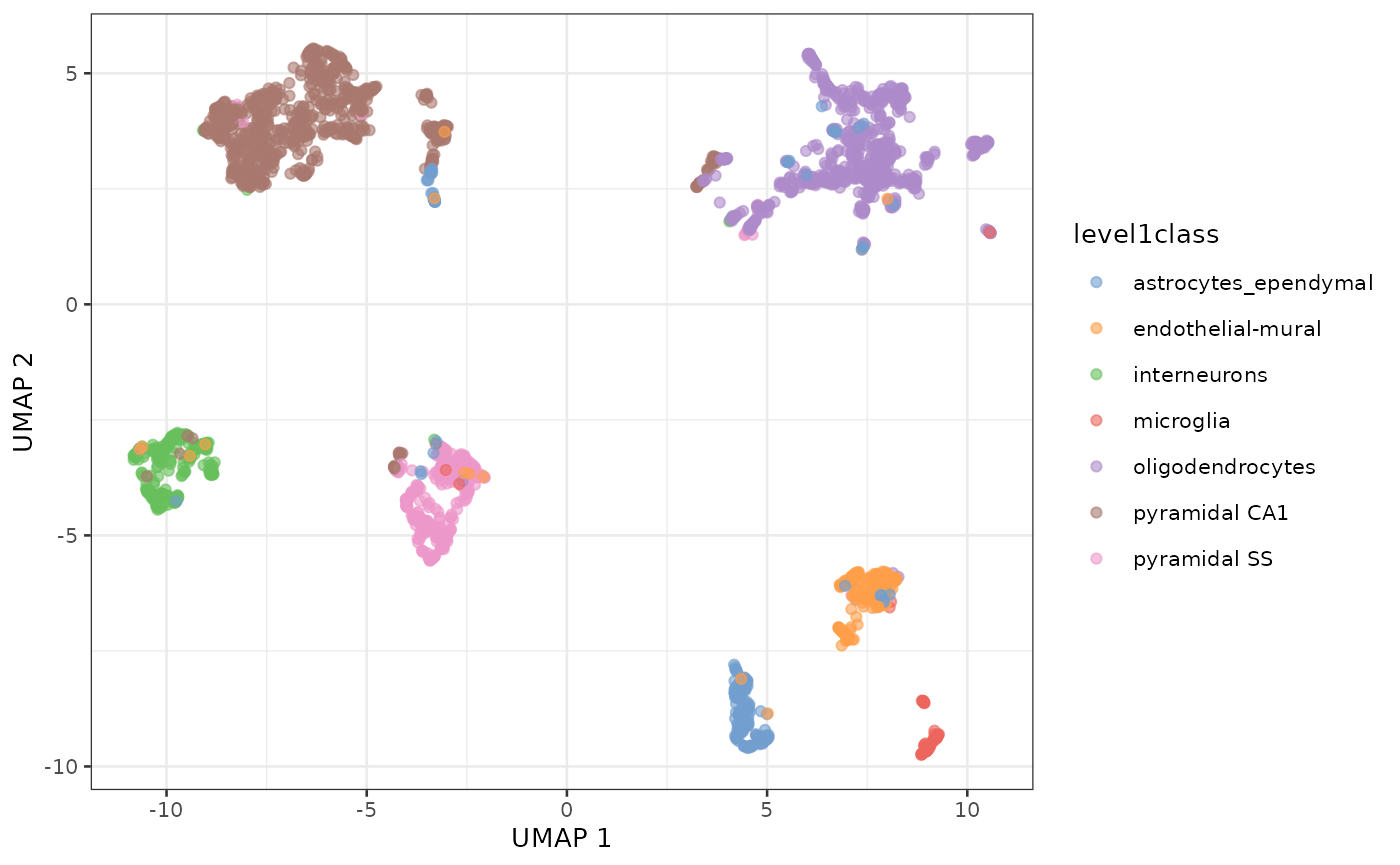
UMAP representation of the Zeisel dataset
tSNE of tissue microarray data
Using default parameters and no log transformation
# We already set up `sce` with `tissue` earlier
sce <- runTSNE(sce, dimred = "PCA")
plotReducedDim(sce, dimred = "TSNE", colour_by = "tissue")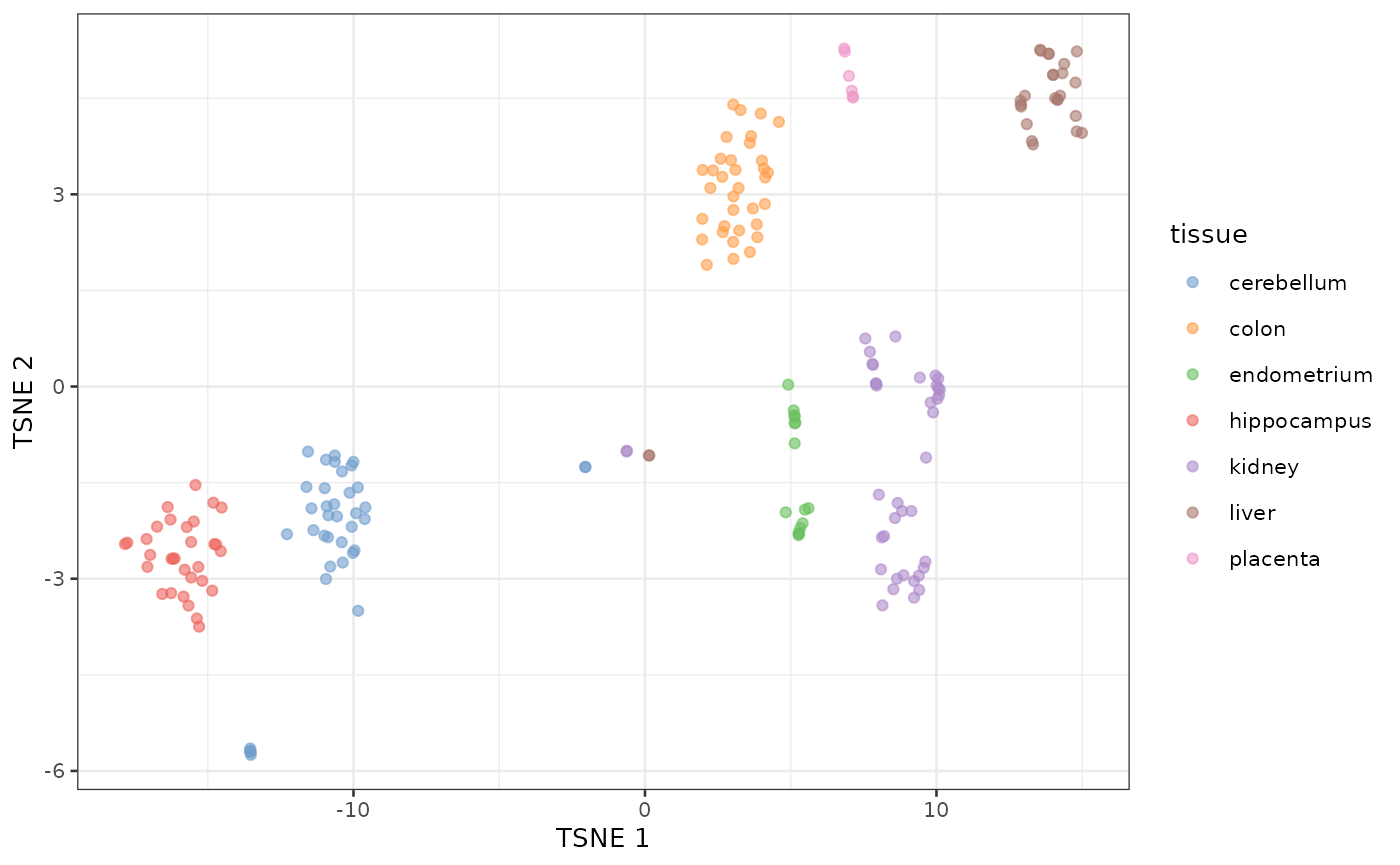
UMAP of tissue microarray data
Also using default parameters and no log transformation
sce <- runUMAP(sce, dimred = "PCA")
plotReducedDim(sce, dimred = "UMAP", colour_by = "tissue")
Summary: distances and dimension reduction
- Note: signs of rotations (loadings) and eigenvectors (scores) can be arbitrarily flipped
- PCA and MDS are useful for dimension reduction when you have correlated variables
- Variables are always centered.
- Variables are also scaled unless you know they have the same scale in the population
- PCA projection can be applied to new datasets if you know the matrix calculations
- PCA is subject to over-fitting, screeplot can be tested by cross-validation
- PCA is often used prior to t-SNE and UMAP for de-noising and computational tractability
Lab exercise
- OSCA Basics: Chapter 4 Dimensionality Reduction
- Optional if you are interested, OSCA Advanced: Chapter 4 Dimensionality reduction, redux
Note about large matrices
- “Stock” R uses an unoptimized
BLASC library for matrix algebra. Large speed gains may be made simply by installing one of (depending on your operating system):- OpenBLAS
- Intel Math Kernel Library
- Apple Accelerate Framework (macOS) (default for MacOS R binaries)