Lab 3: Differential Expression and Double-Dipping
Applied Statistics for High-throughput Biology
lab3.RmdIntroduction
In this lab, we will perform a differential expression analysis using
the airway dataset. We will fit a Negative Binomial
Generalized Linear Model (GLM) using DESeq2. Afterwards, we
will demonstrate the dangers of “double-dipping”—using the data to
define groups, and then testing for differences between those exact same
groups on the same data.
1. The Dataset
We will use the airway package, which provides a
SummarizedExperiment object containing RNA-seq data from
human airway smooth muscle cells. Some cells were treated with
dexamethasone (dex).
library(airway)
library(DESeq2)
library(ggplot2)
data(airway)
se <- airway
se$dex <- relevel(se$dex, ref = "untrt")Take a look at the experimental design:
## DataFrame with 8 rows and 2 columns
## cell dex
## <factor> <factor>
## SRR1039508 N61311 untrt
## SRR1039509 N61311 trt
## SRR1039512 N052611 untrt
## SRR1039513 N052611 trt
## SRR1039516 N080611 untrt
## SRR1039517 N080611 trt
## SRR1039520 N061011 untrt
## SRR1039521 N061011 trt2. Standard DE Analysis (The Right Way)
We will perform differential expression analysis comparing treated vs. untreated samples, controlling for the cell line.
dds <- DESeqDataSet(se, design = ~ cell + dex)
dds <- DESeq(dds)
res <- results(dds, contrast = c("dex", "trt", "untrt"))Let’s look at the distribution of the unadjusted p-values:
hist(res$pvalue, breaks = 50, col = "grey", main = "Standard DE Analysis: trt vs untrt", xlab = "p-value")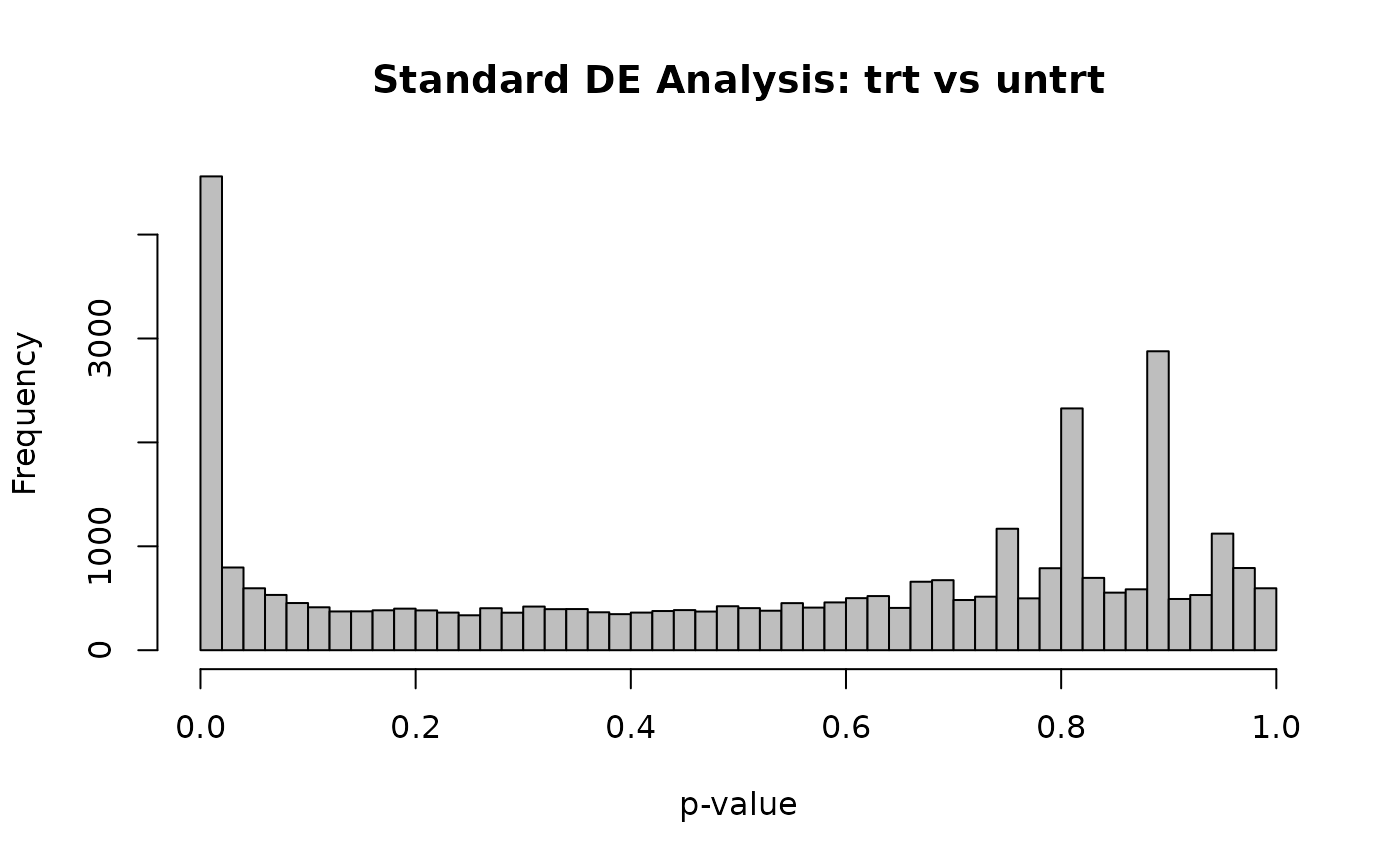
A valid p-value distribution should look roughly uniform under the null hypothesis (features with no true differential expression), with a spike near zero representing the truly differentially expressed features.
3. Demonstrating Double-Dipping (The Wrong Way)
Now we will see what happens when we use the data to define our groups before testing.
Step 3a: Permuted Labels
First, let’s establish a baseline where we know there is no biological signal. We will randomly permute the treatment labels across the samples and run the DE analysis again.
set.seed(123)
dds_perm <- dds
# Randomly shuffle the dex labels
dds_perm$dex <- sample(dds_perm$dex)
# Re-run DESeq
dds_perm <- DESeq(dds_perm, quiet = TRUE)
res_perm <- results(dds_perm, contrast = c("dex", "trt", "untrt"))
hist(res_perm$pvalue, breaks = 50, col = "grey", main = "Permuted Labels", xlab = "p-value")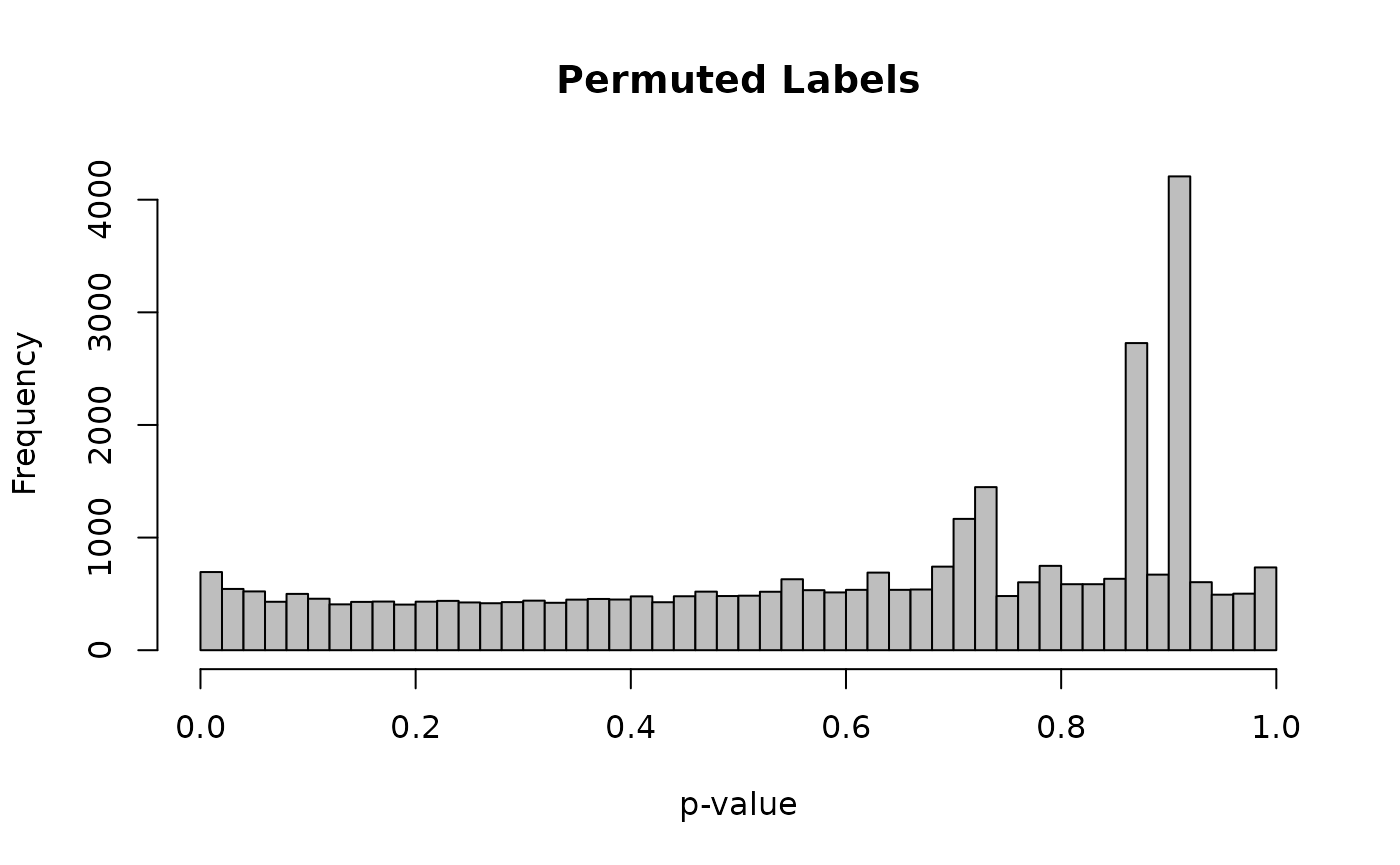
Notice that the spike near zero is gone. The distribution is approximately uniform, which is what we expect when there is no true difference between the groups.
Step 3b: Data-Driven Clustering
Now, imagine we didn’t know the treatment labels. We decide to cluster the samples based on their gene expression profiles, and then test for differential expression between the clusters we just found. This is a classic example of double-dipping.
We will provide pseudocode for how you might accomplish this. Your task is to complete the clustering step.
Recommendation: Before deciding on
clusters, it’s often a good idea to estimate the optimal number of
clusters. You can use a silhouette plot from the cluster
package.
# Pseudocode for Data-Driven Clustering
# 1. Apply a variance-stabilizing transformation to the counts
# vsd <- vst(dds, blind=TRUE)
# 2. Select the top N most variable genes (e.g., top 500)
# rv <- rowVars(assay(vsd))
# select <- order(rv, decreasing=TRUE)[seq_len(500)]
# top_vsd <- assay(vsd)[select, ]
# 3. Transpose the matrix so rows are samples, columns are genes
# ...
# 4. (Optional) Create a silhouette plot to see if k=2 is reasonable
# library(cluster)
# dist_matrix <- dist(...)
# sil <- silhouette(pam(dist_matrix, k=2)$clustering, dist_matrix)
# plot(sil)
# 5. Perform k-means clustering (k=2) on the samples
# set.seed(123)
# km <- kmeans(..., centers=2)
# 6. Assign the resulting cluster labels to a new column in colData
# dds$cluster <- factor(km$cluster)Exercise: Implement the pseudocode above to assign
your samples into two clusters, cluster 1 and
cluster 2, saving it as dds$cluster.
# Write your code here to assign dds$cluster4. Discussion Questions
Explain what happened in Step 3c. Why did the p-value distribution change so drastically when testing between data-driven clusters compared to testing between permuted labels?
-
How might you avoid this problem if you genuinely needed to discover clusters and then test for differences between them? Hint: you can give a brief summary from:
Anna Neufeld, Lucy L Gao, Joshua Popp, Alexis Battle, Daniela Witten, Inference after latent variable estimation for single-cell RNA sequencing data, Biostatistics, Volume 25, Issue 1, January 2024, Pages 270–287, https://doi.org/10.1093/biostatistics/kxac047
Quote from this paper:
“Despite this issue with double dipping, it is common practice. Monocle3 and Seurat are popular packages that each contain functions for (1) estimating latent variables such as clusters or pseudotime and (2) identifying genes that are differentially expressed across these latent variables. The vignettes for these packages perform both steps on the same data (Pliner and others, 2022; Hoffman, 2022). Consequently, the computational pipelines suggested by the package vignettes fail to control the Type 1 error rate. We demonstrate this empirically in Appendix A of the Supplementary material available at Biostatistics online.”