{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
plot(1:10)
plot(10:1)#sweave2rmd
2022-05-09
Levi Waldron (11:27:40): > @Levi Waldron has joined the channel
Levi Waldron (11:27:40): > set the channel description: A project with the goal to convert all Bioconductor package vignettes to R Markdown.
Andres Wokaty (11:28:25): > @Andres Wokaty has joined the channel
Raoul Kamadjeu (17:21:39): > @Raoul Kamadjeu has joined the channel
Paul Villafuerte (22:04:34): > @Paul Villafuerte has joined the channel
2022-05-10
Madelyn Carlson (10:14:19): > @Madelyn Carlson has joined the channel
Madelyn Carlson (10:51:07): > Good Morning, Great to meet y’all via Slack. My name is Madelyn, I am an EPI-BIOS SPH student based in NY and I’m excited to work with you all on this project.@Levi Waldronthank you for the detailed onboarding email yesterday. Per your request, I’m switching to this thread to reply to your message.@Levi WaldronOne question about theBioconductor vignettes Rnw -> Rmd project Trello board- I think that I joined the board but I’m not 100% confident because I’m not showing up as a member. Am I showing up as “joined” on your end? Thank you!
Madelyn Carlson (10:57:34): > Thank you, Jennifer! I just saw that you invited me to the board
Andres Wokaty (10:57:36): > @Madelyn CarlsonI didn’t see you on the board, so I shared it with you. Hopefully that will get you there
Madelyn Carlson (10:57:45) (in thread): > Thank you!
Paul Villafuerte (11:15:36): > hi everyone!
Andres Wokaty (11:38:59): > Hi Paul:slightly_smiling_face:
Andres Wokaty (14:26:06): > I’ll set a meeting for 11:30am-12:30pm ET on Monday. In the meantime, maybe we can do introductions here, like Madelyn did. I’d also like to add if you can speak a little about your technical skill set. If you know you need a little background on something, you can say that too. I just want to start circulating information sooner.
Andres Wokaty (14:30:36): > I’m Jen. My pronouns are they/them. I’m a developer in Levi’s lab and also work on the Bioconductor build system, where I work on Linux, Mac, and Windows machines that run Bash, Powershell, R, and Python. I’m still new to R so I still think in Python:disappointed:I like documenting and troubleshooting things!
2022-05-12
Madelyn Carlson (09:56:21): > Thanks for sending the meeting invite for Monday, Jen. In terms of my technical skill set, I have experience w/ R and SAS from both school and an external research project but I’m not as familiar w/ other programming languages like Python or Bash. I’m interested in learning how to use Python & have been meaning to take a course on it to get basics down. My technical skill set is relatively green. One reason I’m excited about this position is to gain more experience in R, which will be helpful as I transition out of my current job/ field of work experience, which is in grant management. My pronouns are she/her. > > I’m looking forward to getting to know y’all more!
2022-05-16
Andres Wokaty (11:32:37): > Meeting for today:https://meet.google.com/ftp-trea-igo - Attachment (meet.google.com): Meet > Real-time meetings by Google. Using your browser, share your video, desktop, and presentations with teammates and customers.
2022-05-24
Andres Wokaty (16:38:30): > Thanks for meeting Monday. I selected a few Rnw files to start work on. Please assign yourself one athttps://github.com/orgs/Bioconductor/projects/2/views/1and get started. Would everyone be able to meet again at the same time next Monday May 30 11:30-12:30 ET? Levi will be able to join us this time. We can talk about the workflow as well as any issues you ran into. In the meantime, slack me on the channel for conversion issues so that we can learn from each other.
2022-05-25
Andres Wokaty (13:20:00): > @Madelyn CarlsonIs the recording available yet for our meeting on Monday? I just wanted to make sure@Paul Villafuertegot a chance to see it. > > And I want to check if everyone is available for next Monday (see above)@Levi Waldron@Raoul Kamadjeu
Madelyn Carlson (13:31:46): > @Andres WokatyI believe I sent you the recording link in a private Slack DM on Monday. I’ll copy and paste it in this thread so that everyone can access it. > > In terms of next Monday, I am available between 11:30-12:30 ET. & I just claimed on the of Rnw files from the link you sent. Will Slack you with any questions I have been now and when we meet!
Madelyn Carlson (13:32:12): > Here is the recording from our meeting on Monday, 5/23:https://us02web.zoom.us/rec/share/mj5BaEYJjgPj87tdg_7p7y_C47TIBoxVSc0WtteQ_XlKmVxCEB8p0CfzQUDOfanV.aZ9bOQO7STQa_yIL
Andres Wokaty (13:34:51): > @Madelyn CarlsonThank you!
Madelyn Carlson (13:35:08): > Of course! Np:blush:
Paul Villafuerte (13:36:59): > i do remember seeing it earlier this week. Thanks@Madelyn CarlsonI’ll watch this afternoon
2022-05-26
Andres Wokaty (09:55:45): > Hi@Madelyn Carlson,@Raoul Kamadjeu@Paul Villafuerte@Levi WaldronI forgot Monday will be a holiday in the US. Would you be available Wednesday June 1 sometime between 9am - 11 ET work for everyone?
Madelyn Carlson (09:56:35): > On June 1, I’m free from 10-11AM ET
Paul Villafuerte (09:56:54): > 9-10 is good for me. Available again anytime after 1130
Madelyn Carlson (09:57:06): > I could make 9-10 work too
Raoul Kamadjeu (10:23:24): > I am on a different time zone, everything after 12 NY time works for me.
Andres Wokaty (10:49:45): > Should we do 11:30 - 12:30 again then?
Madelyn Carlson (10:52:04): > 11:30-12:30 works for me! On the 1st, I’m free all morning and early afternoon until 1PM ET, so I’m flexible
2022-05-31
Raoul Kamadjeu (05:49:12) (in thread): > Hi Jennifer. Where are the files located. I thought I could just clone them on my local machine, but they are not in a git repo. sorry, this probably has something to do with my still embryonic understanding of Git.
Andres Wokaty (09:45:17) (in thread): > No worries. If you want to do a screenshare before we meet tomorrow, I have time today.
Raoul Kamadjeu (14:05:14) (in thread): > Hi Jennifer. This repository. Where is the GitHub Repo link for it?
Raoul Kamadjeu (14:06:20) (in thread): > the Repo Sweave2Rmd - File (PNG): image.png
Andres Wokaty (14:32:49) (in thread): > https://github.com/Bioconductor/graph
Andres Wokaty (14:35:31) (in thread): > All bioconductor packages are undergithub.com/Bioconductor, like our documentation repohttps://github.com/Bioconductor/Sweave2Rmdand the Bioconductor packages that we’ll be converting Sweave files to Rmd.
Raoul Kamadjeu (14:41:53) (in thread): > Thank you Jennifer, now been able to clone it locally.
2022-06-07
Raoul Kamadjeu (07:51:41): > I Jennifer. I converted a file. How do I submit it for review?
Andres Wokaty (09:49:41): > @Paul Villafuerte@Raoul KamadjeuLet’s walk through the pull request tomorrow so that we all know how to do it. I’ll send the meeting invite for wed 12-1.
Madelyn Carlson (09:50:19): > Sounds perfect. Thank you!
Raoul Kamadjeu (10:30:28): > Super.!
2022-06-08
Raoul Kamadjeu (12:08:25): > Is the meeting happening. There is no one on Zoom
Madelyn Carlson (12:08:51) (in thread): > Madelyn Carlson is inviting you to a scheduled Zoom meeting. > > Topic: Madelyn Carlson’s Personal Meeting Room > > Join Zoom Meetinghttps://us02web.zoom.us/j/7142141473Meeting ID:714 214 1473One tap mobile+16468769923,,7142141473# US (New York)+13126266799,,7142141473# US (Chicago) > > Dial by your location+1 646 876 9923US (New York)+1 312 626 6799US (Chicago)+1 301 715 8592US (Washington DC)+1 669 900 6833US (San Jose)+1 253 215 8782US (Tacoma)+1 346 248 7799US (Houston)+1 408 638 0968US (San Jose) > Meeting ID:714 214 1473Find your local number:https://us02web.zoom.us/u/kb6nlD3ReM
Madelyn Carlson (12:08:58) (in thread): > New Zoom link
Madelyn Carlson (14:35:11): > FYI@Andres Wokaty@Paul Villafuerte@Raoul KamadjeuHere is the meeting recording from this afternoon: > > Meeting Recording:https://us02web.zoom.us/rec/share/qt5CSLuHINdsZhss-XPIawkY1heeoyqsPGz05JOHUKnbLEjQ2wvAxYx60cm-oOw.300RQAT4qJLMVt6L
Andres Wokaty (14:55:25): > @Raoul Kamadjeu@Paul Villafuerte@Madelyn CarlsonI’ve made a few issues inour sweave2rmd reposo that we can start documenting our project. They’re brief in description, but we want to document what we’re doing now so that we can refine later and share it with others. Beyond converting some files to Rmd ourselves–and there are some 900+ files to convert–we want to this to involve into a community project that’s friendly to Bioconductor newcomers so keep this in mind as you write. You’re welcome to pair program/write together or take a single issue. I will try to give some feedback on your PR by next week and if possible aim for another Monday or Wednesday meeting.
Madelyn Carlson (14:58:45) (in thread): > sounds great re: meeting again next week. I think Monday works best for me but Wednesday would also be fine. Times available: 10-11, 11:30-12:30, 4-5. On Wednesday, I’m free 10-11, 12-1 and 1-2
Madelyn Carlson (14:59:58) (in thread): > Will likely have questions in the near future about best ways to document the process
2022-06-15
Andres Wokaty (12:03:09): > @Paul Villafuerte@Levi WaldronMeeting today:https://us05web.zoom.us/j/9280937474?pwd=OURIR1MvL2E2Y0RBMmFKYmZmTHFqdz09.@Levi Waldronif you’re still under deadline, you can skip!
Andres Wokaty (12:41:54): > @Raoul Kamadjeu@Madelyn Carlson^
Madelyn Carlson (12:42:07): > thank you!
2022-06-22
Madelyn Carlson (11:21:31): > Do we already have a Zoom link set up for our 12o’clock? If not, here is a link: > > Join Zoom Meetinghttps://us02web.zoom.us/j/85694664732
Andres Wokaty (12:03:25): > @Paul Villafuerte@Raoul Kamadjeu^
Madelyn Carlson (14:05:24): > Hi@Andres Wokaty(No rush getting to this message btw!) I just finished a first pass at the changes you suggested post-pull request and attempted to push the new file (createReposHtml-Rmd). These are the commands I used to do so: > 1. git commit -m "createReposHtml to Rmd-post edits" > 2. git push origin createReposHtml-Rmd > Although it seemed like there were no errors in my terminal after submitting the second command, the output did not give me a URL to the GitHub page (to complete the pull request) like it did the last time I rangit push origin. I rangit push origin createReposHtml-Rmdagain and the message “Everything up-to-date” came up. I’m worried that the changes I made to the R Markdown file (e.g., removing the extra carriage returns) won’t be reflected in what you can see from your end. Any advice?
Madelyn Carlson (14:58:22): > @Andres Wokaty@Raoul Kamadjeu@Paul VillafuerteHere is the link to meeting recording from June 22 check-in: > > Meeting Recording:https://us02web.zoom.us/rec/share/RS1gVnOUQF5MUF4L-MzrE6fP5-lZ7xAHOSqSTB-zqbR_AvFglDsw_z8psTbUEtV7.ahRyph_xa6ze24fD
2022-06-24
Raoul Kamadjeu (16:12:52): > Thank you. I took the week off and was off-line. Back on my desk on Monday.
2022-07-06
Madelyn Carlson (11:59:34): > hi all, I can’t tell if there is already a Zoom link set up for 12, but if not, here is one:https://us02web.zoom.us/j/85694664732
Andres Wokaty (12:00:28): > thank you:slightly_smiling_face:
Madelyn Carlson (12:02:29): > Whoops, haha. I just accidentally ended the Zoom room. Should be open if we all click the link again
2022-07-11
Andres Wokaty (23:40:16): > I wanted to bring up the genefilter Rmd that@Paul Villafuerteis working on because it’s a good example of what kind of decisions we should make when things get hairy. If you look at the pdf athttps://bioconductor.org/packages/release/bioc/vignettes/genefilter/inst/doc/independent_filtering_plots.pdf, you see the two graphs in Figure 1 that appear side to side. In the text, they’re referred to as ‘left’ and ‘right’; however, when we convert the file to Rmd and use html, html will naturally place the plots where they appear in the content (see Rmdhttps://github.com/villafup/genefilter/blob/independent-filtering-plot-rmd/vignettes/independent_filtering_plots.Rmd#L84-L117and compare with image). There might be a base r-way to put them side-by-side (but I don’t know offhand), but we could option 1 add another package dependency for ggplot2 to put them side-by-side or option 2 move the pictures closer together and change the text to refer to the new locations of graphs.@Levi WaldronWhat do you recommend? I think I would lean toward option 2 since I dislike adding more dependencies, although ggplot2 is likely already to be installed. Whatever the case, we should definitely make note if it in our commit message and bring notice to it in our PR message so that it can be evaluated by maintainers. - File (PNG): genefilter_plots.png
2022-07-13
Andres Wokaty (11:59:00): > Shall we do a huddle today@Paul Villafuerte@Madelyn Carlson@Raoul Kamadjeu? Or do we want to record today? I just want to check in and discuss any issues you’re having.
Madelyn Carlson (12:00:05): > In case we want to record any parts of our conversation, we could Zoom? I’m fine with huddle too. In case we want to Zoom for recording, here is a link:https://us02web.zoom.us/j/85694664732
Paul Villafuerte (12:00:48): > i’m good to huddle
Andres Wokaty (12:00:58): > thank you
2022-07-14
Levi Waldron (08:45:20) (in thread): > Either way would work, but probably the simplest solution would be to usepar(mfrow=c(1, 2))within a single code chunk followed by the two plot commands to put the two subplots side-by-side using the same base R commands. Two separate plots with corresponding changes to the text would be fine two, but converting to ggplot2 seems to me the most complex of the available options.
Paul Villafuerte (15:56:05): > so for first option, we would move code chunk in lines 84-87 into code chunk 110-114? that sounds simple enough. Is that ok even though the sweave document doesn’t read in that order?https://github.com/villafup/genefilter/blob/8ffc90d725a5f478d64733821c5bb4f962df3a70/vignettes/independent_filtering_plots.Rmd#L77-L117
Paul Villafuerte (15:56:54): > was playing around with recordPlot() from base R but doesn
Paul Villafuerte (15:56:59): > doesn’t seem to do the trick
2022-07-20
Andres Wokaty (12:30:43): > This is just a reminder that we won’t meet next week. Please enjoy the conference and have a good week!
2022-07-21
Madelyn Carlson (12:02:41): > Thank you for the reminder!
2022-07-29
Wes W (13:25:10): > @Wes W has joined the channel
Wes W (13:25:20): > great talk, excited to help out
2022-07-30
Levi Waldron (06:08:18): > Hi Wes, great!
2022-08-10
Raoul Kamadjeu (12:03:50) (in thread): > Was a link sent?
Andres Wokaty (13:19:47) (in thread): > @Levi WaldronShould we convert all .Rnw files in the vignettes path or only some? Herve suggested inhttps://github.com/Bioconductor/Biostrings/pull/72#issuecomment-1210013272that we don’t need to convert some .Rnw files, like those that function as “quick references”.
2022-08-23
Andres Wokaty (11:22:08): > @Raoul Kamadjeu@Paul VillafuerteI scheduled a meeting at our usual time tomorrow but if you can’t attend, we can meet individually this week.
2022-08-24
Andres Wokaty (15:09:41): > @Raoul Kamadjeu@Paul VillafuerteI have time Thursday 2-4pm or anytime Friday.
2022-08-25
Paul Villafuerte (11:00:47): > any works for me. whatever works best for you guys
Andres Wokaty (13:50:25): > Let’s meet tomorrow individually when you have time. You can slack me and I can do a huddle or zoom. I’ll be available from anytime except at 10-10:30.
2022-08-31
Andres Wokaty (10:26:23): > @Raoul Kamadjeu@Madelyn Carlson@Paul VillafuerteLet’s check in if you have time today since we skipped last week.
2022-09-01
rohitsatyam102 (09:47:31): > @rohitsatyam102 has joined the channel
Madelyn Carlson (15:15:04): > I’m happy to introduce@rohitsatyam102, who attended BioC2022 and is interested in volunteering with Sweave2Rmd. Re: your questions in our other thread,@rohitsatyam102, here is ourGit pagethat speaks to the project and how it works. If you navigate to vignettes > contribute, you can see guidance on ways to contribute to the project and step-by-step on how we convert sweave docs to Rmd. Feel free to drop more question in this thread. Flagging@Andres Wokaty, who’s our lead (and goes by Jen, btw). Excited to meet you@rohitsatyam102via Slack!
rohitsatyam102 (15:21:24) (in thread): > Nice to join you guys on this. Which one should I will read the vignette and get back to you.
Madelyn Carlson (15:23:34) (in thread): > Amazing! I would suggest reading through the contribute page first and potentially taking a look at our project board (linkedhereand on the Git page hyperlinked in my first message)
Andres Wokaty (15:29:09) (in thread): > Great to have you! We also have a github page version of that repository that’s a bit easier to read athttps://bioconductor.github.io/sweave2rmd/. Let us know if you’d like help picking your first document. - Attachment (bioconductor.github.io): sweave2rmd > Website for Bioconductor community contributors to organize around converting Sweave vignettes to Rmd.
rohitsatyam102 (15:30:42) (in thread): > The GitHub page is soothing to eyes. Thank you. I read it and do I get to pick up a package of my own choice or will one of you assign?
rohitsatyam102 (15:33:15) (in thread): > I would like to take EnrichmentBrowser.
rohitsatyam102 (15:51:13): > Sorry, I didn’t start with my introduction. I am Rohit Satyam and I am a visiting student at King Abdullah University of Science and Technology, Saudi Arabia. I did my major in Bioinformatics from JMI India. I consider myself as intermediate coder in R (I know package development and Shiny App development) but communicating results in reproducible and interactive way has always been my favorite task when I code. I know Bash, Python (basic), Nextflow workflow development (basic) as well, but I am not very good at version control using GitHub. My pronouns are he/him. I always wanted to give back to a community that has always supported my needs as an informatician by providing support and guidance, and I wish to reciprocate in whatever way I can.
Andres Wokaty (16:00:15) (in thread): > Welcome! This project is great for getting to learn git as we use it a lot to communicate and share code.
Andres Wokaty (16:01:40) (in thread): > Sounds good! The first step is to contact the maintainer and ask if they’ll accept the future pull request. The maintainer is pretty friendly and active on Github as well.
2022-09-02
Madelyn Carlson (15:53:17) (in thread): > (Echoing Jen) it’s great to meet you@rohitsatyam102and to learn a bit about your background. Feel free to reach out if you have any Qs about contacting the maintainer (re: our other thread).
2022-09-03
rohitsatyam102 (11:02:25) (in thread): > I actually tried to reach the maintainer a day ago. I haven’t heard from him yet. I emailed viagmailID not institutional (it will expire in a month) and I think since the developer has Harvard Email, it will redirect my email to spam, isn’t it? Should I open it as an issue on his github? Also, how do I editthis pageto update that I contacted the maintainer?
Andres Wokaty (14:32:16) (in thread): > I updated the project page. It might be helpful to make an issue athttps://github.com/lgeistlinger/EnrichmentBrowser/issues. The maintainer, Ludwig, is pretty active on github and on the community slack. If you don’t get a response in the next week or two, I can try to reach out to him too. I would say there’s a high likelihood that he’ll accept the request, so you can keep working on it.
rohitsatyam102 (15:10:56) (in thread): > So should I wait for his reply or start working on Vignette conversion?
2022-09-04
Andres Wokaty (12:11:16) (in thread): > I think it’s okay to start in this case.
2022-09-06
rohitsatyam102 (07:25:59) (in thread): > Okay. I will start from today.
Andres Wokaty (10:51:01): > If everyone’s okay, we can skip our weekly meeting Wed. Sept 7 since it’s a short week. If anyone has questions, needs a review, etc., let’s share this on the channel.@Paul Villafuerte@Raoul Kamadjeu@Madelyn Carlson
Paul Villafuerte (10:53:27) (in thread): > Welcome@rohitsatyam102!
Madelyn Carlson (10:58:04): > Sounds good!
2022-09-07
Andres Wokaty (13:42:21): > @Madelyn Carlson@Paul Villafuerte@Raoul KamadjeuCould I ask for your help in preparing for Sweave2Rmd as a contribution project for Outreachy in the event that we’re accepted? I would like us to identify a few “easy” and “medium” conversions in terms of difficulty and put them on our project boardhttps://github.com/orgs/Bioconductor/projects/2/. I’ve added a column called Difficulty so that we can label the conversion. The trello athttps://trello.com/b/nJHqzR1j/bioconductor-vignettes-rnw-rmd-projecthas a more complete list of Rnw files to choose from. If you find any in the community-high priority list, we should contact the maintainer for the OK so that Outreachy applicants only need to focus on the conversions. This effort won’t be wasted if we don’t participate in Outreachy as it will still be helpful for future Sweave2Rmd volunteers.
Andres Wokaty (13:52:17): > I also wanted to ask your opinions as I am considering if we could possibly open this to Outreachy applicants who do not know R. I could ask them as a task to complete an R tutorial first then do an “easy” conversion.
Madelyn Carlson (13:59:15): > Sure thing! What kind of timeline are we working with to update the “Difficulty” column? & Re: your second message, I think it would be interesting to open it up to Outreachy applicants who do not know R. That is in theme with Sweave2rmd being an opportunity for learning. Also, it could increase our reach
Andres Wokaty (14:05:40): > Thank you. I meant to give a deadline. We need to submit the project by Sept. 23th, but it would be nice to at least 10 listed bySept. 19so I can focus on the workflow/write up for Outreachy. We can continue adding after that as the Outreachy application period doesn’t start until October.
Paul Villafuerte (14:19:36): > What would you consider difficult vs easy. Or should we use our judgment?
Paul Villafuerte (14:20:18): > Would you consider the one with the side-by-side plots medium?
Andres Wokaty (14:26:32): > That’s a good question. You can use your judgement. I would lean on the side-by-side plots being medium since you might need to know a little extra R to handle formatting issues.
2022-09-09
rohitsatyam102 (22:47:10): > Hi everyone. I stumbled upon an error while converting.Rnwfile to.md. Idk why it expects\end{document}in middle of the document when it’s already present at the end? > > Error at "EnrichmentBrowser.tex" (line 350, column 13): > expecting \end{document} > \end{center} > ^ >
2022-09-12
Andres Wokaty (10:03:31) (in thread): > We’ve found that sometimes during this conversion that pandoc’s having an issue converting usually a chunk of text. If you know the problem area, you might want to temporarily remove that chunk so that you can convert the rest of the document. You can then add that chunk back in, which you should convert manually (since pandoc didn’t help).
Andres Wokaty (10:04:51) (in thread): > If you get stuck on it, I’m happy to meet for a Slack huddle and we can walk through it together.
rohitsatyam102 (10:06:32) (in thread): > Okay let me try it one more time with removing that code chunk and if it continues to be an issue let’s meet then.
2022-09-14
rohitsatyam102 (00:19:59) (in thread): > It got resolved by deleting that section.
rohitsatyam102 (00:23:31): > Hi I wish to ask how the words highlighted in red in vignette PDF need to be taken care of? Should I just use backticks? - File (PNG): image.png
Andres Wokaty (08:51:25) (in thread): > Yes, backticks should be fine
Raoul Kamadjeu (11:52:39) (in thread): > Good to know. If the Rnw file ia too long, you can use the utility I created to directly convert tx to Rmd
Raoul Kamadjeu (11:53:18) (in thread): > https://kamadjeu.com/projects/Tex2Rmd/
Raoul Kamadjeu (11:54:11) (in thread): > You need to do some manual tweaking still, and work on the blank spaces in the yaml header
rohitsatyam102 (12:48:00) (in thread): > I believe this is the first proper Tex to Rmd convertor that I came across. Why don’t you make it as a separate R package?
2022-09-19
Madelyn Carlson (09:55:25) (in thread): > GM ~ FYI I added 10 vignettes to ourGit project boardfor our future Outreachy volunteers (see bottom of board). All new vignettes are marked as “easy” or “medium” difficult level. URLs for vignette PDFs and/or the Bioconductor landed pages can be as comments if you click on the title of the vignette in Git. For the vignettes from packages we need to contact maintainers, I emailed or Git DMed (whatever the term is for Git, haha) all maintainers. We’ve already heard back from 1, others are pending. Please let me know how else I can help w the Outreachy application!@Andres Wokaty
2022-09-20
Madelyn Carlson (11:12:54): > @Madelyn Carlson has joined the channel
2022-09-21
rohitsatyam102 (21:02:56) (in thread): > Hi guys. I am swamped with work until second week of October. Is that fine? I am running a bit slow on my volunteering part
Andres Wokaty (22:48:01) (in thread): > No, worries!
2022-09-26
Paul Villafuerte (13:53:33): > I’ve come across a document where the pdf on the landing page is different from the rnw file. > > PDF**** ****here****:****The one I’m working on has this in the Rnw: > > RNW: > > We will use the info data to compare quality measures between > novel (i.e., not in dbSNP) and known (i.e., in dbSNP) variants > and the variant type present in the file. Variants with > membership in dbSNP can be identified by using the appropriate > SNPlocs package for hg19.<<examine_dbSNP>>=``library(SNPlocs.Hsapiens.dbSNP.20101109)``rd <- rowRanges(vcf)``seqlevels(rd) <- "ch22"``ch22snps <- getSNPlocs("ch22")``dbsnpchr22 <- sub("rs", "", names(rd)) %in% ch22snps$RefSNP_id``table(dbsnpchr22)And thePDF: > > We will use the info data to compare quality measures between novel (i.e., not in dbSNP) > and known (i.e., in dbSNP) variants and the variant type present in the file. Variants with > membership in dbSNP can be identified by using the appropriate SNPlocs package for the > hg19 genome (GRCh37).library(SNPlocs.Hsapiens.dbSNP144.GRCh37)``vcf_rsids <- names(rowRanges(vcf))``chr22snps <- snpsBySeqname(SNPlocs.Hsapiens.dbSNP144.GRCh37, "22")``chr22_rsids <- mcols(chr22snps)$RefSNP_id``in_dbSNP <- vcf_rsids %in% chr22_rsids``table(in_dbSNP)
Andres Wokaty (14:22:07) (in thread): > There are a few reasons why this might happen. Your repository has gotten behindhttps://github.com/Bioconductor/VariantAnnotation. If you look at your repositoryhttps://github.com/villafup/VariantAnnotation, you’ll see a message saying that it’s 1 commit behind, so you’re looking at the previous version of the document that we can see athttps://github.com/Bioconductor/VariantAnnotation/blob/de7f434f13d2b5a333ead0ba260b4d953102a53d/vignettes/VariantAnnotation.Rnw. You want to update your local and remote repositories, using the method we discussed a few weeks ago to add the Bioconductor VariantAnnotation as a remote, fetch it, and rebase your master branch on it. > > # Let's switch back to our master branch > git switch master > # Add Bioconductor/VariantAnnotation as a remote named 'upstream' > git remote add upstream git@github.com:Bioconductor/VariantAnnotation.git > # Download references from upstream > git fetch upstream > # Replay the difference of changes from upstream's master branch to your local master branch > git rebase upstream/master > > You can always check the changes by examining thegit logbefore and after the operations. The .Rnw should be updated after that. You can either create a new branch for your changes or rebase on the branch you made for conversion. For example, if your new branch wasvariantannotation-rmd, you would do > > git switch variantannotation-rmd > git rebase master >
rohitsatyam102 (15:40:07): > Hello everyone. How do I add paper references in RMD? Currently they are added as[@Chiaretti04]and this is rendered as(S et al. 2004). Should I keep it that way? or do i need to make i a hyperlink like we usually add links to a word in markdown using[]()syntax.
Andres Wokaty (17:35:10) (in thread): > This is a good question as we haven’t come across it yet. Could you make the rendered text a hyperlink?
rohitsatyam102 (20:06:41) (in thread): > Below is the attached file. - File (HTML): EnrichmentBrowser.html
Andres Wokaty (20:09:59) (in thread): > They didn’t render as links?
rohitsatyam102 (20:10:46) (in thread): > No
Andres Wokaty (20:11:33) (in thread): > If you commit your work and push your branch up to your github repo, I can take look at the code.
rohitsatyam102 (20:12:47) (in thread): > Yes, I will try doing that tomorrow because this is the final knit. I will learn that and do it.
Andres Wokaty (20:13:10) (in thread): > No worries, let me know if you have questions, even how to do that.
rohitsatyam102 (20:14:57): > So in the default guidelines of new packages that are being submitted, is it now mandatory to use RMD generated HTML vignette? or people are still using Sweave format?
Andres Wokaty (20:28:40) (in thread): > Good point! The documentation doesn’t require Rmd format and mentions .Rnw as an option:https://contributions.bioconductor.org/docs.html. I’ll see if we can try to ‘recommend’ the Rmd format in the documentation to avoid future .Rnw documents. - Attachment (contributions.bioconductor.org): Chapter 12 Documentation | Bioconductor Packages: Development, Maintenance, and Peer Review > Package documentation is important for users to understand how to work with your code. 12.1 Bioconductor documentation minimal requirements: a vignette in Rmd or Rnw format with executable code…
rohitsatyam102 (20:35:02) (in thread): > Yes because there was this wonderful package that was submitted in 2021 I think but the pdf version vignette had commands trimmed which makes is unpleasant and difficult to follow.
2022-09-27
Andres Wokaty (20:28:54) (in thread): > Thanks to your comment, we made .Rmds recommended:https://contributions.bioconductor.org/docs.html. - Attachment (contributions.bioconductor.org): Chapter 12 Documentation | Bioconductor Packages: Development, Maintenance, and Peer Review > Package documentation is important for users to understand how to work with your code. 12.1 Bioconductor documentation minimal requirements: a vignette in Rmd or Rnw format with executable code…
2022-09-28
rohitsatyam102 (11:31:25) (in thread): > Okay guys I need some rescuing. I tried to push the changes but there is this nast.gitdirectory that’s preventing to push the changes. I deleted it and now it’s angry with the error. I always thought this directory was useless. > > fatal: not a git repository (or any parent up to mount point /) > Stopping at filesystem boundary (GIT_DISCOVERY_ACROSS_FILESYSTEM not set). >
Andres Wokaty (12:54:01) (in thread): > That’s an important directory. Without it, git thinks it’s not part of a repository. If it’s deleted and you can’t undo it, such as by restoring from your trash, I would rename this directory, such as toEnrichmentBrowserOld. I’dgit clone git@github.com:Rohit-Satyam/EnrichmentBrowser.gitagain. Then I’d create my new branch in the new cloned directory withgit switch -c enrichmentbrowser-rmd. I would then copy over the files I changed fromEnrichmentBrowserOldto the newEnrichmentBrowser. Then you can make commits again.
2022-09-29
rohitsatyam102 (09:33:19) (in thread): > I tried this:https://gist.github.com/Rohit-Satyam/8b71a68db7d5cb3083018aabe533e036What am I missing? > Update1: I was able to generate the token that’s now the default way of pushing changes. Can you check if I did it correctly:https://github.com/Rohit-Satyam/EnrichmentBrowser/tree/enrichmentbrowser-rmd
Andres Wokaty (11:55:02) (in thread): > Yes, you did it correctly! Just one note: when you clone, you don’t have to dogit initas that’s usually for creating a new repository.
Andres Wokaty (11:57:00) (in thread): > When you commit, don’t commit the.htmlproducts. We just want the.Rmd. I’ll take a closer look at it later and try to get back to you tomorrow or early next week.
2022-10-04
Paul Villafuerte (16:38:03): > Having a problem with VariantAnnotation. MycommitLine 196 creates a plot with ggplot:ggplot(metrics, aes(x=RSQ, fill=in_dbSNP)) + geom_density(alpha=0.5) + scale_x_continuous(name="MaCH / Thunder Imputation Quality") + scale_y_continuous(name="Density") + theme(legend.position="top")The plot takes a very long time to generate, but my output is completely different from the output in the original pdf. Both are attached. > > Be warned that the plot takes about 15 minutes to generate. To reproduce you might want to trim the dataset:metrics <- metrics[c(1:200),] - File (PDF): VariantAnnotation_3.pdf - File (PNG): From PDF
Paul Villafuerte (16:39:54): > I think the problem is with the fill argument in ggplot:fill=in_dbSNP.If you remove it, it gets a little closer to the graph from the original.
2022-10-08
Phylis Atieno (13:10:56): > @Phylis Atieno has joined the channel
Phylis Atieno (13:15:31): > Hi everyone! My name is Phyllis an Outreachy applicant. I’m so excited to be here and I’m applying to contribute to the ‘Conversion of Sweave vignettes to R markdown’ project. Will be reaching out with questions along the way :)
Madelyn Carlson (13:22:25) (in thread): > Welcome, Phyllis! Great to Slack-meet you. And yes, feel free to drop in any questions about the conversation process in this channelortheOutreachythread. Happy to answer any Qs you have.
Haleema Sadia (13:59:24): > @Haleema Sadia has joined the channel
Haleema Sadia (14:02:10): > Hi Everyone, > I am Haleema khan and I am an Outreachy applicant. I am aiming to contribute to the “Convert Sweave vignettes to R Markdown” Project and glad to be a part of the community. Currently working on setting up the environment.
Phylis Atieno (14:03:58) (in thread): > Thank you Madelyn!
2022-10-09
rohitsatyam102 (10:13:27): > @Andres Wokatywere you able to have a look at my code. Is it fine to ask theEnrichmentBrowserdeveloper to pull changes?
rohitsatyam102 (10:24:12): > Welcome@Phylis Atienoand@Haleema Sadia. Please have a look atSweave2Rmdwebsite. > > Steps to convert the .Rnw files to Rmd are simple and are described here. You first rename the .Rnw file to .tex and upload the .Rnw filehere. And then you save the results as .Rmd (Credits to@Raoul Kamadjeu). Then you manually fix the .Rmd formatting. You can read more atcontribute page - Attachment (bioconductor.github.io): Contribute > Learn how to contribute to our project.
Phylis Atieno (10:29:40) (in thread): > Thank you@rohitsatyam102!
Haleema Sadia (10:37:04) (in thread): > Thanks alot!
Andres Wokaty (11:43:54) (in thread): > Yes, please see the pull request for my comments as well as Ludwig’s:https://github.com/lgeistlinger/EnrichmentBrowser/pull/44
Chizi E (13:22:52): > @Chizi E has joined the channel
rohitsatyam102 (15:57:31) (in thread): > Do I just need to click on resolve conversation?
darmimessele seyoum (18:52:21): > @darmimessele seyoum has joined the channel
darmimessele seyoum (18:54:08): > Hello everyone. My name is Darmi Seyoum. I wanted to apply to join this team. My pronouns are she/her. I will await further instructions from the mentors
Haleema Sadia (21:33:43) (in thread): > @rohitsatyam102I am new to R markdown and trying to add code chunks to my rmd. > I have a question/confusion: > For a sample code to load it needs to be executed in the rmd file and if yes then should the rmd file contain all the import commands or there is a way to link it to the code file. > E.g if I add a chunk that say load(data) as an example of code in the main .R file then I get a “not found” error.
YuNan Guan (22:51:28): > @YuNan Guan has joined the channel
YuNan Guan (23:00:42): > Hi everyone:) My name is YuNan Guan, the applicant of Outreachy. I’m applying to work on sweave2rmd project. My pronouns are she/her.
2022-10-10
rohitsatyam102 (03:09:35) (in thread): > You have to install the package whose Vignette you are updating and load the Library first. Yes there must be a code chunk to load all necessary libraries and dependencies. Which package you are working with?
Halimat Chisom (03:22:42): > @Halimat Chisom has joined the channel
Halimat Chisom (03:24:56): > Hello everyone, > My name is Halimat, an Outreachy applicant. I’m applying to contribute to the ‘Conversion of Sweave vignettes to R markdown’ project. My pronouns are She/Her. > I’m looking forward to working with you. I just started setting up, so I don’t have any questions for now. > > Thank you
Madelyn Carlson (10:03:38) (in thread): > Welcome, Haleema! Greet to meet you. Let us know if you have any questions related to setting up the environment.
Madelyn Carlson (10:08:50) (in thread): > Welcome, Darmi! Great the meet you via Slack. To get started, feel free to check out our Sweave2Rmd page if you haven’t already:https://bioconductor.github.io/sweave2rmd/In it, there are instructions onhow to begin a conversation. Also, here is alist of vignetteswe are in the process of converting as well as ones waiting to be converted. Please Slack if you have any questions! Happy to help. - Attachment (bioconductor.github.io): sweave2rmd > Website for Bioconductor community contributors to organize around converting Sweave vignettes to Rmd. - Attachment (bioconductor.github.io): Contribute > Learn how to contribute to our project.
Madelyn Carlson (10:10:32) (in thread): > Welcome, Halimat! Great to meet you. As you continue setting up & prepping for the conversions, feel free to drop any questions in this thread or in the Outreachy channel. Happy to help.
darmimessele seyoum (10:54:36) (in thread): > Nice to meet you Madelyn. I will check out the links you have sent me and reach out if I have any questions.
Madelyn Carlson (10:55:17) (in thread): > Perfect!
Andres Wokaty (11:55:49): > Hi Outreachy applicants. Please make sure that you’re do the contribution tasks labeledoutreachy dec22in thehttps://github.com/Bioconductor/sweave2rmd/issues. I’ve labeled them so that their set up (labelled Prep Work) walks you through the process incrementally to do tasks.
Haleema Sadia (13:04:12) (in thread): > I am currently working with the biobase package and trying to convert the sweave to rmd. > > I have successfully generated the rmd file and the code chunks as well but when I try loading the project, it gives errors.
Haleema Sadia (14:29:25): > Hi, > I am trying to understand how knit works? > I wrote an rmd to write a vignette and loaded the project then loaded the relevant methods to make the code chunks work and it was fine until i tried to knit it. It threw the errorobject not foundI did some google and found out that my current loaded data is in my workspace but knit creates a new environment where I dont have anything loaded so I have to manually load it in my code chunks. Is it how its supposed to work? I mean if before all the example I have to call all the methods in the RMD file then isnt it repeating lengthy codes? Or I should add the codes and turn the input code view to not visible. > any pointers would be helpful. Thanks
Haleema Sadia (15:10:49) (in thread): > I was able to resolve the issue by adding the load all command before my code chunks and turning visibility to run only.
Andres Wokaty (16:01:35) (in thread): > Please leave them as I will use them as my guide when I review the vignette again. I’ll marked them resolved.
2022-10-11
Chinwe (00:34:02): > @Chinwe has joined the channel
Chinwe (00:43:45): > Hello everyone:wave:My name is Chinwendu Nwazojie.You can call me Chinwe. I am applying to work on this project as an outreachy intern for the Dec 2022 round. > My pronouns are she/her/hers. I am looking forward to contributing to this project.:slightly_smiling_face:
Chinwe (01:10:32) (in thread): > Thank you for this@rohitsatyam102just reading through the channel and I came across this. It will come in handy soon:grinning:
Chinwe (01:23:06) (in thread): > Okay, thanks for this. I went through the issue tracker and it seems that all the projects labelledoutreachy dec22has been taken but with this message I now understand that it is a task for all outreach applicants. > > > I suggest it be pinned to the chat so new applicants can find it.:slightly_smiling_face:@Andres Wokaty
Atuhurira Kirabo Kakopo (08:59:24): > @Atuhurira Kirabo Kakopo has joined the channel
Haleema Sadia (11:00:54): > Hi@Andres WokatyI created a PR yesterday for under Sweave2rmd for Biobase package. Who do I need to tag there?
2022-10-12
Haleema Sadia (02:16:48): > Hi all, > I am trying to convert a.texfile to.mdbut getting the error ” Unexpected end of file” > After searching so far I have understood that it might be due to some syntax error in the LaTex but is there a way to trace the error. The file is long and I am not sure what to exactly look for.
Haleema Sadia (02:18:30) (in thread): > Hi@rohitsatyam102. just tried using the mentioned tool. It gives an error whenever I submit a.texfile as follows: - File (PNG): Screenshot 2022-10-12 at 11.18.01 AM.png
krunal gamit (03:22:23): > @krunal gamit has joined the channel
rohitsatyam102 (04:13:17) (in thread): > did you change the file extension from . Rnw to .tex via command line? If you are still getting error, try the command line way using pandoc.
rohitsatyam102 (04:15:13) (in thread): > > # Copy the file.Rnw to file.tex since pandoc understands .tex files > cp file.Rnw file.tex > # Convert file.tex to Markdown and save as file.md > pandoc -s file.tex -o file.md > # Rename the Markdown file to R Markdown > mv file.md file.Rmd >
rohitsatyam102 (04:18:16) (in thread): > If you get an error remove that chunk from MD file. Usually the line at which error occurs is given in the error.
Akwamfon Eventus (07:11:56): > @Akwamfon Eventus has joined the channel
Akwamfon Eventus (07:23:52): > Hi Everyone<!here>, > Hello Mentor@Andres WokatyI’m Akwamfon Eventus (Him/He), an applicant for the Dec22 Outreachy Open Source Internship. > I am applying to contribute on this project. I wish to contribute to the “sweave2rmd” project. > Great to be here. (edited)
Madelyn Carlson (11:19:23) (in thread): > Welcome, Akwamfon! Great to meet you.
Madelyn Carlson (11:20:09) (in thread): > Welcome, Chinwendu! Great to meet you:wave:
Madelyn Carlson (11:21:01) (in thread): > Perfect, thank you for updating your work-around here for other folks who may run into the same issue.
Haleema Sadia (12:05:08) (in thread): > I tried it using the tool you shared and also via pandoc commands like above. > I constantly getUnexpected end of file"It is giving error at last line and nothing is there except the document end tag.
Haleema Sadia (12:07:39) (in thread): > Tried both.
Haleema Sadia (13:45:16) (in thread): > Says error on line 689. I cannot understand because its end of document. - File (PNG): Screenshot 2022-10-12 at 10.43.39 PM.png - File (PNG): Screenshot 2022-10-12 at 10.44.40 PM.png
2022-10-13
Haleema Sadia (02:09:08) (in thread): > I was able to resolve the issue by thoroughly reading the .tex file. > The file was constantly pointing at end of file where as shown above nothing was wrong. > When I started reading and analysing tags i found the character$in a formula that was causing the issue. I removed it , converted to .Rmd and added it in the file again at relevant place. > > Another helpful thing was that I noticed the colour of some characters like< < / > > -etc which was different in the .tex file with errors vs a normal valid .tex file. Might not be something special but it helped me track the exact place where colour started changing and I found the special character.
Haleema Sadia (02:09:38) (in thread): > thanks alot@rohitsatyam102for helping out!
rohitsatyam102 (09:19:42) (in thread): > First and foremost. I am blow by how meticulously spotted the formatting mistakes. Is there a code or something that you use to do this or it was manual inspection? Besides, I took your suggestion on reformatting the references according to BMC Bioinformatics format. Is this the result you expecting? - File (PNG): image.png
rohitsatyam102 (09:21:10) (in thread): > Also, now that I have made changes to the.Rmd, should I overwrite theenrichmentbrowser-rmdbranch or create a fresh branch like somethingenrichmentbrowser-rmd-v0.1?
rohitsatyam102 (09:22:26) (in thread): > Also, I would encourage@Haleema Sadiaand@Phylis Atienoto have a look at suggestions made by Jennifer to avoid mistakes made by me.
rohitsatyam102 (09:33:27) (in thread): > In addition, I learned that the in-text citations can be referenced usinglink-citations: TRUE > > bibliography: > - refs.bib > link-citations: TRUE > csl: bmc-bioinformatics.csl >
rohitsatyam102 (09:37:33): > I was wondering if there is a way to reduce the blank space present on the left and right side of the HTML vignette. It feels too empty and misuse of space and makes the vignette longer. Is there a logical reason to keep that space empty? - File (PNG): image.png
Andres Wokaty (15:49:19) (in thread): > I manually inspected the files.:sweat_smile:You should commit your changes to the same branch and push them so they get attached to the pull request then ask again for a review so we can check off what has been complete. I was expecting the output you got for the bibliography. Let’s Ludwig if this is sufficient in the PR comments. > > Also, if you have time, would could you write how you did the bibliography in our Troubleshooting FAQ athttps://github.com/Bioconductor/sweave2rmd/blob/main/vignettes/troubleshooting-faq.Rmdwith a PR. I think it would be really helpful.
Andres Wokaty (19:02:32): > I think you could make it so that the table of contents doesn’t float (so it’s just at the top) to reduce horizontal scrolling but otherwise the space is fine.
rohitsatyam102 (19:08:04) (in thread): > Sure. I would love to.
2022-10-14
Phylis Atieno (01:41:18) (in thread): > Thank you@rohitsatyam102will surely do
Chinwe (10:47:44): > Hello everyone, Good day. > > Please for the first task that has to do with creating a package. Please can someone point me to an article or anything to help me get started? I have installed R and R studio and forked and cloned the package template but do not know how to proceed from there. Please any help or explanation will do:pleading_face:
Andres Wokaty (11:04:30) (in thread): > We can talk about this in the session today:slightly_smiling_face:
2022-10-15
Chinwe (02:48:37): > Hello everyone, Please I am trying to import the bugsigdb and I get this warning message every time.@Andres Wokaty - File (PNG): image.png
Andres Wokaty (09:35:59) (in thread): > I think this is a bug because the slashes are mixed and you’re using Windows. You should be able to remove the file manually withfile.remove()and with the path to the file using all\slashes.
Andres Wokaty (09:41:27) (in thread): > After removing the file, you should be able to run it again.
Andres Wokaty (09:45:58) (in thread): > I see someone already reported the issue athttps://github.com/waldronlab/bugsigdbr/issues/32
2022-10-17
Maria Fiorio (14:18:52): > @Maria Fiorio has joined the channel
2022-10-18
Chinwe (00:11:12) (in thread): > thanks Jen
Halimat Chisom (07:39:11): > Hi@Andres Wokaty@Madelyn CarlsonDo I have to leave a comment on the second issue for assignment before I can start working on any project?
Halimat Chisom (08:16:56) (in thread): > Nevermind… I think I figured it out. > The graph package page on BiocViewshttps://bioconductor.org/packages/devel/bioc/html/graph.htmldoesn’t have theURLof the repo underDetailsas I expected. > > I’ve requested an assignment on the second issue. - Attachment (Bioconductor): graph (development version) > A package that implements some simple graph handling capabilities.
Madelyn Carlson (09:49:30) (in thread): > @Halimat ChisomGM! To clarify, you requested to be assigned tograph/vignettes/GraphClass.Rnw, right?
Halimat Chisom (09:49:59) (in thread): > Yes
Madelyn Carlson (09:50:12) (in thread): > Done!
Madelyn Carlson (09:50:47) (in thread): > FYI I assigned you using your GitHub username
Halimat Chisom (09:51:54) (in thread): > Thank you:slightly_smiling_face:
rohitsatyam102 (10:11:23) (in thread): > @Andres Wokatycommitted changes as suggested in FAQ and pushed the new RMD file toenrichmentbrowser-rmdbranch.
Chinwe (17:02:10): > Hello,@Halimat Chisomare you done with the fist and second contribution tasks? If yes please can you put me through I am stuck.
Andres Wokaty (17:42:17) (in thread): > If you can share more about where you are stuck, I can give you some guidance.
Haleema Sadia (22:40:49): > Hi, > Issue: Unable to generate scatter plot for a vignette. Any insights are welcome.(See below for details) > I am working on the matchprobes vignette. > cc@Andres WokatyActual result: - File (PNG): Screenshot 2022-10-19 at 7.40.34 AM.png
Haleema Sadia (22:41:21) (in thread): > Expected: - File (PNG): Screenshot 2022-10-19 at 7.41.05 AM.png
2022-10-19
Halimat Chisom (00:12:53) (in thread): > I’m done with the first task. Feel free to ask your questions here. The mentors will respond@ChinweWhat have you done so far?
Chinwe (05:05:17) (in thread): > Okay, I am currently following a text article on how to create a package. During our last call with Jen, they told me to add an R folder and vignette file. > > I do not know what the content will be. So following the article it says I should run a command > > usethis::create_package("C:/Users/Shannon.Pileggi/Desktop/ralph") > > I ran the command while replacing the path with my own path for my preferred location on my computer. I got this error. - File (PNG): image.png
Chinwe (05:05:43) (in thread): > I have been stuck here for some days now.
Chinwe (05:06:23) (in thread): > I also do not know how to use the template from the github account that we were supposed to use,
Chinwe (05:07:17) (in thread): > this is link to the article I am usinghttps://www.pipinghotdata.com/posts/2020-10-25-your-first-r-package-in-1-hour/ - Attachment (PIPING HOT DATA): PIPING HOT DATA: Your first R package in 1 hour > Tools that make R package development easy
Chinwe (05:08:31) (in thread): > And this is a screenshot from my Rstudio. - File (PNG): image.png
Halimat Chisom (08:12:51) (in thread): > Hi Chinwe, > > I’m not sure what that line does, but for the first task, all you have to do is follow the instructions. > So, I’ll ask, have you forked and cloned the package template repo to your device? That should be your first step. > If yes, then on your RStudio, navigate to that particular folder. > In the folder, there’s an R folder with an R script for hello word. You can either edit that by writing a new function that uses bugsigdbr to perform some tasks, or you delete the R script and write a new script with bugsigdbr. > I’ll stop here because I’m not sure how far you’ve gone. > > > > If you’ve done all this, you can mention that in your response.
Chinwe (09:58:51) (in thread): > I have cloned the package template
Andres Wokaty (10:16:42) (in thread): > The packageTemplate is essentially an incomplete Biconductor package. As Halimat mentioned, you should follow the instructions to add the function and a vignette in the correct places. It’s also helpful to look at other existing packages, likehttps://github.com/waldronlab/bugsigdbr, to see what the structure looks like.
Chinwe (10:41:58) (in thread): > This is the content of my R script now
Chinwe (10:42:02) (in thread): > > #' Hello World > #' > #' @return character > #' @export > #' > #' @examples > #' helloWorld() > ngr.obesity.feces <- function() { > newDB <- importBugSigDB() > ngr.obesity.feces <-subset(newDB, `Location of subjects` == "Russia" & Condition == "obesity" & `Body site` == "feces") > } >
Andres Wokaty (10:45:02) (in thread): > That’s a good start. Remember, your function should take some inputs. You should also edit the comments above.
Chinwe (10:46:53) (in thread): > ok
Chinwe (10:49:30) (in thread): > I just created a folder named vignette and a file newDB .Rmd file in it
Andres Wokaty (10:50:41) (in thread): > The folder should be namedvignetteswith an s. It sounds like you’re on the right path now.
Chinwe (11:40:55) (in thread): > ``{---} > title: "MicroB" > author: "Chinwendu Nwazojie" > date: "Sys.Date()`” > output: rmarkdown::html_vignette > vignette: > > % > % > % > — >
Vignettes are long form documentation commonly included in packages. Because they are part of the distribution of the package, they need to be as compact as possible. The
html_vignetteoutput type provides a custom style sheet (and tweaks some options) to ensure that the resulting html is as small as possible. Thehtml_vignetteformat:
- Never uses retina figures
- Has a smaller default figure size
- Uses a custom CSS stylesheet instead of the default Twitter Bootstrap style
Vignette Info
Note the various macros within the
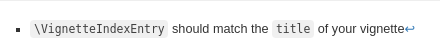
vignettesection of the metadata block above. These are required in order to instruct R how to build the vignette. Note that you should change thetitlefield and the\VignetteIndexEntryto match the title of your vignette.Styles
The
html_vignettetemplate includes a basic CSS theme. To override this theme you can specify your own CSS in the document metadata as follows:output: rmarkdown::html_vignette: css: mystyles.cssFigures
The figure sizes have been customised so that you can easily put two images side-by-side.
You can enable figure captions by
fig_caption: yesin YAML:output: rmarkdown::html_vignette: fig_caption: yesThen you can use the chunk option
fig.cap = "Your figure caption."in knitr.More Examples
You can write math expressions, e.g. \(Y = X\beta + \epsilon\), footnotes1, and tables, e.g. using
knitr::kable().Also a quote using
>:“He who gives up [code] safety for [code] speed deserves neither.” (via)](https://twitter.com/hadleywickham/status/504368538874703872))) ```
Chinwe (11:41:17) (in thread): > This is the content of the file in the vignettes folder
Andres Wokaty (11:42:00) (in thread): > Your vignette should explain how to use the function you wrote.
Chinwe (11:42:32) (in thread): > okay let me look at the repo you shared earlier.
Haleema Sadia (15:43:15) (in thread): > Hi@Andres Wokatywere you able to take a look or if you have any pointers that’d be great
Andres Wokaty (15:45:58) (in thread): > Could I ask you to push your branch to your repository and I can look at the code there?
Haleema Sadia (16:06:19) (in thread): > sure
Haleema Sadia (16:25:27) (in thread): > I think its resolved. I was trying to generate it via code. but when I saw the folder a png got created with the plot which i have added as image. > I will open a PR for feedback.
2022-10-20
Heather Turner (03:09:59): > @Heather Turner has joined the channel
Adeoye Beatrice (10:02:16): > @Adeoye Beatrice has joined the channel
rohitsatyam102 (21:57:38): > I would like to start with my second contribution if@Andres Wokatyyou are satisfied with EnrichmentBrowser. I will assignggbioto myself.
2022-10-21
rohitsatyam102 (12:09:23): > Okay. The maintainer of the ggbio is unaware about Rmds and he is asking what’s the point of this project. I would love to say that I hate Pdf vignettes:joy:since the code is often clipped in them, and they are difficult to navigate, but I am sure you guys have a better argument prepared for such question. Please see here:https://github.com/lawremi/ggbio/issues/166
Andres Wokaty (12:22:44) (in thread): > This is a good point. I think we should probably add the rationale to the README. I summarized it here:https://docs.google.com/presentation/d/11PX1CWperCET_oDMM43KSTIhwsuTtuqAO1kgY7Pq96w/edit#slide=id.g168379e359d_0_5
Andres Wokaty (12:26:26) (in thread): > I made an issue for it athttps://github.com/Bioconductor/sweave2rmd/issues/37. I think the maintainer gave a good reason, which is that they don’t really remember Sweave/Latex:wink:
Andres Wokaty (12:35:06) (in thread): > Can I ask you to leave open that issue you made making the request? You can automatically close it with the corresponding PR by writingClose #166(where 166 is the issue number) in the PR’s comments when the PR is merged. This way we nicely tie the conversation/issue with the pull request.
2022-10-22
rohitsatyam102 (11:13:52) (in thread): > @Andres WokatyCan you help?
rohitsatyam102 (12:31:55): > @Andres WokatyWhat do I do if a Vignette has Chapters?
rohitsatyam102 (12:34:08) (in thread): > I am thinking to treat chapters as sections and replace word “Chapter” with word “Section”
Phylis Atieno (14:22:19): > Hi everyone, can someone please help me on this. When converting a .Rnw vignette to R markdown, can this code\begin{verbatim} > bplapply(fls[1:3], FUN, BPPARAM = MulticoreParam(), param = param)be converted to R markdown like this > > {verbatim} > bplapply(fls[1:3], FUN, BPPARAM = MulticoreParam(), param = param) >
rohitsatyam102 (15:47:21) (in thread): > I am afraid the answer is NO. The website provided for the Rnw to Rmd conversion is experimental and can only do so much and therefore you have to manually delete\begin{verbatim} >. You can useCtrl+Fshortcut to find and replace such occurrences, if they occur at multiple places in the file.
rohitsatyam102 (15:52:06): > For some weird reason, the BiocStyle is not properly placing margin notes and making printing them as footnotes. For example, I copied this from BiocStyle github > and pasted it my vignette to see how it is being rendered and they are being put at the end of the sessionInfo rather than being displayed beside the location where they are cited: > > label^[for cross-references to work the chunk > label may only contain alphanumeric characters (a-z, A-Z, 0-9), slashes (/), or > dashes (-)] > - File (PNG): image.png - File (PNG): image.png
{kind=link}
{kind=link}
2022-10-23
Phylis Atieno (07:07:18) (in thread): > Thank youu@rohitsatyam102
2022-10-24
Andres Wokaty (10:21:53) (in thread): > Sorry, I had thought I had responded. I think the white space is fine. This is just the default formatting from BiocStyle, which has an optimal width of 80. You could alternatively just have the table of contents just at the top rather than floating at the right, which would reduce space.
Andres Wokaty (10:43:12) (in thread): > I think it might be better to omit this because I think both might be a little problematic when trying to format it with R Markdown. When they’re formatted in R Markdown, it will just show up as “1 Getting Started” then the text. However you choose to handle it, I would note with an explanation it for the maintainer/reviewers in the PR’s comments.
rohitsatyam102 (11:07:39) (in thread): > okay. Thanks
2022-10-26
Andres Wokaty (09:29:44) (in thread): > Can we see more of what is surrounding this code because it’s not anything obvious to me.
2022-10-28
Haleema Sadia (11:14:55): > Hi@Andres WokatyI hope you are well. > While filling the final application form , which community questions should we answer?
Andres Wokaty (11:28:09) (in thread): > I seem to be having trouble accessing it at the moment, so I am reaching out to Outreachy. I’m glad you’ll be submitting a final application.
Haleema Sadia (11:30:56) (in thread): > Looking forward to it :’)
Haleema Sadia (11:31:07) (in thread): > I hope it gets resolved soon!
2022-10-31
Halimat Chisom (01:44:20): > Hi@Andres Wokaty@Madelyn CarlsonCan someone help with installing this package? I can’t find it on the Bioconductor page and this conversion won’t work unless it’s installed. - File (PNG): library.png
{kind=link}
Halimat Chisom (01:59:40) (in thread): > Another question about the final application: should we mention the first contribution of creating R package even though there was no PR or merging?
Andres Wokaty (09:42:17) (in thread): > @Haleema SadiaI think I might not have access to change or see the questions and I don’t want to delay anyone’s submission. Regarding the application, try to answer all the questions as best as possible.@Halimat ChisomSince I think I can’t see the final application until you submit, I am going to suggest that if you’re allowed to mention more than one then include it. If you’re only allowed to mention one contribution, pick the one you merged or made a PR for.
Haleema Sadia (09:45:53) (in thread): > Here you go:https://bioconductor.org/packages/release/data/experiment/html/RNAseqData.HNRNPC.bam.chr14.html - Attachment (Bioconductor): RNAseqData.HNRNPC.bam.chr14 > The package contains 8 BAM files, 1 per sequencing run. Each BAM file was obtained by (1) aligning the reads (paired-end) to the full hg19 genome with TopHat2, and then (2) subsetting to keep only alignments on chr14. See accession number E-MTAB-1147 in the ArrayExpress database for details about the experiment, including links to the published study (by Zarnack et al., 2012) and to the FASTQ files.
Halimat Chisom (09:46:40) (in thread): > Thanks@Haleema Sadia
Haleema Sadia (09:47:31) (in thread): > So currently the box does not say any question in specific but says Answers to community questions. > As per past interns, the orgs give them questions internally and they are supposed to add their answers there.
Halimat Chisom (09:50:23) (in thread): > It’s an optional question though. How about a screenshot? Would that work?
Andres Wokaty (09:51:42) (in thread): > Sure. Thank you both!
Halimat Chisom (09:52:33) (in thread): > Here… - File (PNG): comm_questions.png
{kind=link}
Andres Wokaty (09:54:09) (in thread): > Thanks. I will speak with the mentor of our other project and post the questions in the#outreachyby tomorrow. I appreciate your help :)
2022-11-03
darmimessele seyoum (20:38:49) (in thread): > Hey Madelyn I tried to take a task from the list but it is not allowing me…
darmimessele seyoum (20:39:11) (in thread): > Can u help me
2022-11-04
Madelyn Carlson (12:12:58) (in thread): > Hithankyoufor reaching out!Whichvignettefromtheprojectboarddoyouwanttoclaim?Icanassignittoyouontheboard.
darmimessele seyoum (12:13:54) (in thread): > I am okay with any leftover vignette as long as it’s moderated
Madelyn Carlson (12:19:26) (in thread): > Great I assigned you to genefilter/vignettes/howtogenefinder.RnwLinkto Bioconductor landing page. Please send your Git username and I can update the name assigned to the vignette. - Attachment (Bioconductor): genefilter > Some basic functions for filtering genes.
darmimessele seyoum (12:22:58) (in thread): > My github user name is Darmimese
darmimessele seyoum (12:23:05) (in thread): > Do what should I do next??
darmimessele seyoum (12:23:14) (in thread): > Could you tell me the next steps?
Madelyn Carlson (22:36:29) (in thread): > Apologies for the delay, I was at a conference today and wasn’t able to switch back online. Re-circulating information about next steps after a vignette is assignedhere.
2022-11-08
Andres Wokaty (12:42:30): > @Paul VillafuerteSorry, I ‘overruled’ parts of your review. Martin suggested inhttps://github.com/Bioconductor/sweave2rmd/issues/33that we could give contributors credit, which I am 100% behind, but the documentation hasn’t been updated yet.
Paul Villafuerte (12:50:05): > got it. Was there anything else? I don’t see anything on github, or maybe I don’t know where to check.https://github.com/Bioconductor/S4Vectors/pull/111
Andres Wokaty (12:59:24) (in thread): > I haven’t finished going through that review. It probably take me a few weeks to go through them.
2022-11-16
Vince Carey (13:41:06): > @Vince Carey has joined the channel
2022-11-29
rohitsatyam102 (08:08:07) (in thread): > I am so sorry I was juggling between projects and I almost forgot about this. Yes here is the portion: > > Overview is a good way to show all events at the same time, give overall summary statiics for the whole genome label^[for cross-references to work the chunk > label may only contain alphanumeric characters (a-z, A-Z, 0-9), slashes (/), or > dashes (-)] . >
2022-11-30
Andres Wokaty (23:19:18) (in thread): > No worries! I see what you mean. It’s apparently because we have different versions of pandoc:https://github.com/Bioconductor/BiocStyle/issues/101. Because these are links, it’s okay they go to the bottom of the page. They’ll likely be rendered differently (hopefully as margin notes) on the build system. - Attachment: #101 Margin notes and other style > Cześć Andrzej! > > I am having a styling issue and would be great if you could help me. In an older version of the BiocStyle, the references and footnotes were going to the margin, which is exactly what I want:
> image > > Now, when I take your code and try to recreate it, I get the footnotes at the bottom of the document.
> image
> image > > Is it possible to force it to the margin? > > Here is the output of sessionInfo() on the system on which this document was compiled: > > > > ## R version 4.1.3 (2022-03-10) > ## Platform: x86_64-pc-linux-gnu (64-bit) > ## Running under: Ubuntu 22.04.1 LTS > ## > ## Matrix products: default > ## BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.10.0 > ## LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.10.0 > ## > ## locale: > ## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C > ## [3] LC_TIME=de_DE.UTF-8 LC_COLLATE=en_US.UTF-8 > ## [5] LC_MONETARY=de_DE.UTF-8 LC_MESSAGES=en_US.UTF-8 > ## [7] LC_PAPER=de_DE.UTF-8 LC_NAME=C > ## [9] LC_ADDRESS=C LC_TELEPHONE=C > ## [11] LC_MEASUREMENT=de_DE.UTF-8 LC_IDENTIFICATION=C > ## > ## attached base packages: > ## [1] stats graphics grDevices utils datasets methods base > ## > ## other attached packages: > ## [1] BiocStyle_2.22.0 > ## > ## loaded via a namespace (and not attached): > ## [1] bookdown_0.27 digest_0.6.29 R6_2.5.1 > ## [4] jsonlite_1.8.0 magrittr_2.0.3 evaluate_0.15 > ## [7] highr_0.9 stringi_1.7.6 rlang_1.0.2 > ## [10] cli_3.3.0 rstudioapi_0.13 jquerylib_0.1.4 > ## [13] bslib_0.3.1 rmarkdown_2.14 tools_4.1.3 > ## [16] stringr_1.4.0 xfun_0.31 yaml_2.3.5 > ## [19] fastmap_1.1.0 compiler_4.1.3 BiocManager_1.30.18 > ## [22] htmltools_0.5.2 knitr_1.39 sass_0.4.1 > > >
{kind=link}
{kind=link}
{kind=link}
2022-12-02
Beryl Kanali (14:43:10): > @Beryl Kanali has joined the channel
2022-12-13
Marcel Ramos Pérez (13:57:30): > @Marcel Ramos Pérez has joined the channel
2022-12-20
Madelyn Carlson (10:32:17): > Great talking with y’all this AM. Creating a list of agenda items for next Sweave2Rmd meeting to keep them top of mind for myself: > * Discuss method for contacting maintainers (preference for opening issues, cc’ Jen on any emails) > * Strategize standardize ways to make the Sweave2Rmd project board work effectively for our work. How can it be structured to better reflect real time changes? > cc@Beryl Kanali@Paul Villafuerte@Andres Wokaty
2022-12-29
Madelyn Carlson (12:59:54): > hi hi ~ In the Sweave2Rmd Trello board pre-triage list, the following package/vignettes were not found in theBioconductor build/check report list. Flagging@Andres Wokatyas this could mean that they will be removed from the pre-triage list > * AffyCompatible/vignettes/MAGEAndARR.Rnw > * AffyCompatible/vignettes/NetAffxResource.Rnw > & the following vignettes had a build error. These were sorted into their appropriate stacks in the Trello board. > * AneuFinder/vignettes/AneuFinder.Rnw > * ArrayExpress/vignettes/ArrayExpress.Rnw > I have only finished packages that begin with “A” as of now so there may be additional items added to this list later on. I’ll can this post as a running list.
Andres Wokaty (14:01:35) (in thread): > Thanks for working on this. It looks like AffyCompatible was deprecated. Anything that’s deprecated will be removed from Bioconductor, so we can just put them inwon’t fixwith a note they’re deprecated. I moved them there now with a note.
Madelyn Carlson (14:02:20) (in thread): > Perfect, thank you!
2023-01-04
Madelyn Carlson (14:07:58): > Happy New Year, all! If anyone is interested in starting a new Sweave2Rmdconversion, here are a few “hot off the press” (i.e., the pre-triage Trello stack) vignettes, maintained by the core team that are ready to convert and merge: > * Category/vignettes/Category.Rnw > * Category/vignettes/ChromBand.Rnw > * ensemblVEP/vignettes/ensemblVEP.Rnw > * gpls/vignettes/gpls.Rnw > You can find these & more corresponding info per each vignette in ourSweave2Rmd project boardunder status: “todo” - Attachment (bioconductor.github.io): Contribute > Learn how to contribute to our project.
2023-01-05
rohitsatyam102 (00:42:13) (in thread): > I would like to take ensemblVEP. I am just finishingggbiodocumentation (couple of lines left). However, I can see ggbio missing from Sweave2Rmd project board. Can you please add it?
rohitsatyam102 (05:52:56): > @Andres WokatyThe.Rmdfile for ggbio has been updated and is available athttps://github.com/Rohit-Satyam/ggbio/blob/ggbio-rmd/vignettes/realvignettes/ggbio.Rmd. If you are satisfied, I will send them pull request.
Madelyn Carlson (08:18:55) (in thread): > amazing! Thank you@rohitsatyam102. I added you to ensemblVEP on theproject board.@Andres Wokaty-double checking that the ggbio maintainer Michael Lawrence has given us the green light to merge the updated vignette? If not, we/I can send them a quick message.
Andres Wokaty (14:32:00) (in thread): > I think the conversion should be reviewed first.@rohitsatyam102Can you make the pull request and ask me to me review with@jwokaty? On the board, it should be set toIn Review.
Madelyn Carlson (14:57:48) (in thread): > Agreed, re: reviewing prior to requesting merge. For the vignette Rohit converted (from ggbio package), it is not currently listed on the Sweave project board and is community maintained, which might mean that Michael Lawrence hasn’t been contacted yet. Do you think we should reach out to Michael now to ask if they’re interested in the conversion or pause until you’ve finished first review?@Andres Wokaty
Andres Wokaty (16:34:20) (in thread): > @rohitsatyam102contacted Michael and he was receptive:https://github.com/lawremi/ggbio/issues/166. It’s on the board, using the name of the issue. If you hover over the link in the board, you’ll see it’s to ggbio. - Attachment: #166 Vignette Conversion Request > Hi,
> I’m volunteering with Sweave2Rmd and we’re trying to convert all Bioconductor Sweave vignettes into Rmd files. Would you be willing to accept a pull request converting “ggbio: visualization toolkits for genomic data” for the “ggbio package” from one of our volunteers? We’ll take care of converting the file. We’ll ask you to review and accept the pull request. You’ll also need to bump the version number in your DESCRIPTION file so that the build machines will recognize that a new version is available. > > Thanks for your time.
Madelyn Carlson (19:53:21) (in thread): > Whoops, I missed that.My bad! Thank you@Andres Wokatyand@rohitsatyam102!
2023-01-06
rohitsatyam102 (02:01:24) (in thread): > @Andres Wokatypull request generated and @jwokaty tagged.
2023-01-07
rohitsatyam102 (14:45:23): > Having a hard time installing VEP and then connecting it with Rstudio in windows to run VEPFlags function. Any suggestions? Even after installing DBD::mysql, I get this. Installation via mamba isn’t working either. > > WARNING: DBD::mysql module not found. VEP can only run in offline (--offline) mode without DBD::mysql installed[http://www.ensembl.org/info/docs/tools/vep/script/vep_download.html#requirements](http://www.ensembl.org/info/docs/tools/vep/script/vep_download.html#requirements)curl failed (000), trying to fetch using LWP::Simple > ERROR: Failed last resort of using HTTP::Tiny to download[https://raw.githubusercontent.com/Ensembl/ensembl-vep/release/108/modules/Bio/EnsEMBL/VEP/Constants.pm](https://raw.githubusercontent.com/Ensembl/ensembl-vep/release/108/modules/Bio/EnsEMBL/VEP/Constants.pm) >
2023-01-09
Andres Wokaty (11:56:12) (in thread): > What OS are you using? Could you briefly summarize the installation steps you took and led to this error? I haven’t used mamba. But I have installed ensemblVEP before following Bioconductors internal documentation and what’s on ensemblVEP’s github.
rohitsatyam102 (23:15:43) (in thread): > using Windows and trying to install VEP on wsl. Since I see Rstudio picking up wsl in its”Terminal” tab I think it should work.
2023-01-10
Andres Wokaty (09:53:34): > Sorry, I froze
Madelyn Carlson (09:54:04): > All good! Are you able to log back in or would it be easier for connectivity to do Slack huddle?
Andres Wokaty (09:54:26): > Maybe slack huddle
Beryl Kanali (10:17:24): > Hi<!here>Here is a blog I wrote about Bioconductor in general and a little about Sweave2Rmd as part of my Outreachy Internshiphttps://dev.to/berylkanali/think-about-your-audience-with-bioconductor-3ld0 - Attachment (DEV Community 👩💻👨💻): Think About Your Audience (with Bioconductor) > Welcome….again! In this blog, I will explain the Bioconductor project and my experience…
Madelyn Carlson (10:17:43) (in thread): > Thank you for posting!
2023-01-11
rohitsatyam102 (15:29:25) (in thread): > @Madelyn CarlsonI tried VEP installation both on Windows (WSL) and Linux (on separate workstation). I was able to do it successfully in Linux. However, I couldn’t get it to communicate with my Rstudio. I have raised a query regarding same herehttps://github.com/Bioconductor/ensemblVEP/issues/6. I thought I would be able to finish this up within few days but VEP configuration is posing issues. I can however share the vignette. Rmd file if you guys have VEP installed already. If the issue gets resolved timely, its fine, otherwise I have to give up this vignette conversion task. - Attachment: #6 VEPFlags refuses to accept the scriptPath of VEP > I successfully installed VEP in my ubuntu v.20 and added its path to ~/.bashrc as shown below but still I get the error
> .bashrc file content > > > export PATH="$PATH:/home/subudhak/ensembl-vep-release-108" > alias vep="sudo /home/subudhak/ensembl-vep-release-108/vep" > > > Error msg > > > Error in validObject(.Object) : > invalid class “VEPFlags” object: Ensembl >=88 requires vep script (not variant_effect_predictor.pl) > > > When trying to explicitly define the path the error message persists: (even renamed vep to variant_effect_predictor.pl) > > > param <- VEPFlags(flags=list(host="useastdb.ensembl.org"), scriptPath = as.character("home/subudhak/ensembl-vep-release-108/variant_effect_predictor.pl")) > Error in validObject(.Object) : > invalid class “VEPFlags” object: Ensembl >=88 requires vep script (not variant_effect_predictor.pl) >
Andres Wokaty (15:34:27) (in thread): > I’ll take a look at the issue. You can still make a PR. If I’m not able to figure out the issue with VEP, I can take over PR so that we build on your work.
Madelyn Carlson (15:35:21) (in thread): > @rohitsatyam102thank you for the update! I’m sorry that VEP installation is giving you a hard time. Appreciate your next steps, Jen. Happy to support however I can
2023-01-17
Madelyn Carlson (09:02:11): > GM! Re: the comment on our project board for reviewingIndependent filtering plot rmd, comment: “@mcarlsn, did I accidentally assign you or did you take this one?” I’m happy to review it. This is just a FYI
Andres Wokaty (10:44:43): > @Beryl Kanali@Madelyn Carlson@Paul VillafuerteCould you share the output of your.libPaths()?
Paul Villafuerte (10:48:46): > > "/mnt/STORE1/lib/R/bioc-release" "/usr/lib/R/site-library" "/usr/lib/R/library" >
Andres Wokaty (11:00:55): > @Paul VillafuerteIt’s strange that you don’t have a local library
Madelyn Carlson (12:11:01): > > [1] "/mnt/STORE1/lib/R/bioc-release" "/usr/lib/R/site-library" "/usr/lib/R/library" >
Madelyn Carlson (12:11:35): > Mine looks the same as Paul’s : / explains why we’re both getting the same error
Beryl Kanali (12:21:42): > > "/mnt/STORE1/lib/R/bioc-release" "/usr/lib/R/site-library" > "/usr/lib/R/library" >
Andres Wokaty (15:15:38): > @Paul Villafuerte@Madelyn Carlson@Beryl KanaliCould I ask you to try installing a package withBiocManager::installas we did in our meeting today? I updated your permissions and you may want to logout/restart session to see if that changes anything. > > Regarding a local library, I noticed that not everyone has one. If you want one you can set it up. > > I have one set up at/home/jwokaty/R/x86_64-pc-linux-gnu-library/4.2(you can make the directories if you don’t have them) and put the following at the end of my /home/jwokaty/.bashrc:R_LIBS_USER="/home/jwokaty/R/x86_64-pc-linux-gnu-library/4.2". You need to logout and log back in for your .bashrc to take effect. When you do.libPaths(), it should show your local library at the top. This shows library paths it checks in the order it checks for a package.
Marcel Ramos Pérez (15:35:12): > Why not used the centralized repository of packages/mnt/STORE1/lib/R/bioc-release? What errors are you getting@Madelyn Carlson?
Andres Wokaty (15:38:26): > They were getting a permission error when trying to install. I added them to the waldronlab group, so maybe they’ll be able to install now. I just wanted to also provide info if they wanted their own library, too.
2023-01-18
Madelyn Carlson (10:48:35) (in thread): > I see Jen’s response below. Let me know if there’s any other info I can add that would be helpful! Thanks for your help btw
2023-01-19
rohitsatyam102 (07:54:35) (in thread): > Hi guys. Any update on this? Should I share the .Rmd file I made here?
Andres Wokaty (10:57:47) (in thread): > I started looking at this last week but stopped because I’ve been sick a few days. I’ll look again today and try to follow up tomorrow.
2023-01-29
Paul Villafuerte (16:39:32): > I’m working ongpls.pdfand when converted, code blocks are inserted that look like that the sections are to be explicitly not numbered. E.g., the introduction had# Introduction {#introduction .unnumbered}appended once converted. Should we keep this format or convert to numbered sections? > > Also, the last section “Fitting Models to data”is numbered.
2023-01-30
Andres Wokaty (09:56:38) (in thread): > Let’s try to match what is in the original document. Unless it becomes problematic (too much effort) or not possible to represent it in the same way we should follow that. I will try to look at the document today (I am offline right now) so I can understand the problem better.
2023-02-10
rohitsatyam102 (01:48:58): > Hi everyone. I applied for outreachy Internship this February. After how long will I know if I am shortlisted or not?
Andres Wokaty (09:03:20) (in thread): > Outreachy will review your initial application to make sure that you’re eligible. If you’re eligible, you can participate in the contribution period in March. To apply for sweave2rmd, you’ll have to submit a contribution then you can submit your application for the project.
rohitsatyam102 (09:39:29) (in thread): > I have already contributed two package conversions in sweave2rmd. Does that count?
Andres Wokaty (09:40:56) (in thread): > I’m sorry I don’t think it works like that. I think we have to choose based on what’s done during the contribution period.
rohitsatyam102 (13:09:55) (in thread): > Thanks a lot for the info. Really appreciate
2023-02-13
Madelyn Carlson (18:33:10) (in thread): > Amazing to hear! We’ll circulate more information about the contribution period once it’s closer to date
2023-03-07
Sonali Kumari (02:52:19): > @Sonali Kumari has joined the channel
Sonali Kumari (12:15:20): > hello@Andres Wokatyi want to contribute on the projecthttps://github.com/Bioconductor/graph/pull/9as written in the documentation I have to change the status to . Also, write your GitHub username underAssignedand setLast Modifiedto today. but I am unable to do that please help me with it . my github url ishttps://github.com/sonali8434 - Attachment: #9 Converting GraphClass.Rnw to GraphClass.Rmd
Andres Wokaty (12:17:41) (in thread): > Please make a comment in the issuehttps://github.com/Bioconductor/sweave2rmd/issues/63so that the sweave2rmd team can manage these requests. - Attachment: #63 Replace a Sweave Vignette With an R Markdown Vignette > Hello Outreachy applicants! > > Note: This task can be completed by multiple Outreachy applicants. Please choose this as your second (optional) contribution task to the Bioconductor Sweave2Rmd project as it builds upon the familiarity quiz in the Sweave2Rmd first contribution. > > Prep Work > > • Sweave2Rmd Knowledge Check > • Please read our contribution document, which gives a good overview of the project plus the general workflow. In particular, you will be performing the tasks from <https://bioconductor.github.io/sweave2rmd/articles/contribute.html#create-the-r-markdown–rmd-vignette-and-remove-the-sweave-vignette–rnw|Create the R Markdown (.Rmd) vignette and Remove the Sweave vignette (.Rnw)>. > • Install pandoc > > Contribution Task > > Make a pull request replacing a Sweave vignette with an R Markdown vignette. > > 1. Select a vignette from our project board that is labeled ‘Easy’ or ‘Medium’, set to ‘Todo’ and are unassigned. > 2. Write a comment with the path of the vignette you’d like to do–e.g., BiocParallel/vignettes/Introduction_To_BiocParallel.Rnw to @Bioconductor/sweave2rmd. We will write your GitHub username as ‘Assigned’ and set ‘Last Modified’ to today. > 3. Perform the steps in <https://bioconductor.github.io/sweave2rmd/articles/contribute.html#create-the-r-markdown–rmd-vignette-and-remove-the-sweave-vignette–rnw|Create the R Markdown (.Rmd) vignette and Remove the Sweave vignette (.Rnw)>. Note: You do not need to contact the maintainers to work on this set of vignettes as they have either been previously contacted OR they are maintained by the Bioconductor Core Team. > 4. Make sure that you request a review by writing @Bioconductor/sweave2rmd in a comment so that we can review your pull request. For example, @Bioconductor/sweave2rmd please review my contribution for Outreachy. > 5. If you receive any feedback requesting changes, please address them. > > Tip: You can find the PDF you’ll be replacing by visiting the landing page for the package under Documents. You can look up the package at <https://bioconductor.org/packages/devel/BiocViews.html#___Software|Bioconductor Software Packages>.
> Tip: You can read how we <https://bioconductor.github.io/sweave2rmd/articles/contribute.html#review-a-pull-request|review PRs> in the contribution document. Use the checklist to assess if your PR will pass review. > > If your pull request is good, it will be merged into Bioconductor! > > Remember to record your contributions in Outreachy at https://www.outreachy.org/outreachy-may-2023-internship-cohort/communities/bioconductor/convert-sweave-vignettes-to-r-markdown/contributions/.
Sonali Kumari (12:18:42) (in thread): > thank you .
KritikaVerma (12:19:15): > @KritikaVerma has joined the channel
KritikaVerma (12:21:34): > Hii everyone > I am Kritika Verma. I am currently studying IT and Mathematical Innovation with minor in ComputationalBiology. I am an Outreachy applicant for the May cohort. I look forward to learning from all of you. > Thank you
Madelyn Carlson (14:52:26) (in thread): > Welcome Kritika, great to e-meet you!
Madelyn Carlson (14:54:51) (in thread): > @Sonali KumariAlso, unless you’ve done so already, please start with the first contribution task (instructions here) if interested in the Sweave2Rmd project. Once completed, you have the option of starting asecond (optional) contribution taskin which you claim a vignette from the project board and convert it. The first contribution task is designed as a ramp to the second task, which is why we ask that you work on them sequentially. Please let us know if you have questions!:relaxed: - Attachment: #62 Sweave2Rmd Knowledge Check - Attachment: #63 Replace a Sweave Vignette With an R Markdown Vignette
Sonali Kumari (15:02:36) (in thread): > ohk thanks for the information@Madelyn Carlson
Madelyn Carlson (15:02:50) (in thread): > Absolutely!
Monalika Patnaik (15:08:04): > @Monalika Patnaik has joined the channel
Monalika Patnaik (15:24:29): > Hello everyone:wave:Myself Monalika, an outreachy applicant from Delhi, India. would love to make valuable contributions in the project and looking forward to assist and learn from other fellow developers:blush:
Emmanuel Taiwo devbird007 (22:44:18): > @Emmanuel Taiwo devbird007 has joined the channel
2023-03-08
Sweta Kumari (00:02:57): > @Sweta Kumari has joined the channel
Ibizugbe Merit (00:15:30): > @Ibizugbe Merit has joined the channel
Unyime William (01:15:23): > @Unyime William has joined the channel
MaryJones (01:31:57): > @MaryJones has joined the channel
MaryJones (01:36:07): > Hello everyone, Mary Jones, an Outreachy applicant, Bsc IT, South Africa. I want to add my little valuable quota to the projects . I am excited and very happy to learn from the experts and mentors here. Thank you for the opportunity:pray:
MaryJones (01:40:04): > On that note, I have completed some steps of the Contribute article but**** I can’t find the repository for the genefilter package we are meant to fork ****and work with , please may someone kindly advice me on what are instructed to do in a simple way to complete the contribute quiz. Thank you in advance .
Reece (02:20:44): > @Reece has joined the channel
Rukky (02:33:30): > @Rukky has joined the channel
Monalika Patnaik (03:39:59) (in thread): > @Andres Wokaty
Monalika Patnaik (03:40:36): > I also need some assistance too!
KritikaVerma (03:54:08) (in thread): > Nice to e-meet you too!
MaryJones (05:19:49): > @Andres Wokaty: I am sorted, found it and working on it.
Treasure Okafor (05:34:05): > @Treasure Okafor has joined the channel
Treasure Okafor (06:27:01): > Hello everyone, I’m Treasure. Delighted to be here, looking forward to learning, contributing to projects and working with you all:+1:
Treasure Okafor (06:55:11): > Please, I am trying to contribute to a project. I was reading through the instructions, specifically the contribute.rmd in the sweave2rmd repository. > number 2 in claiming the vignette to convert > Select an issue to work on by setting Status to Todo, Last Modified to today, and change Assigned to your GitHub username. > > I couldn’t find my github username, do i still go on with it? > > Also, please is there anyone working with the same project. Please I need guidance:pray:
Monalika Patnaik (07:12:17) (in thread): > 1. Hey@Treasure OkaforWelcome to the community ..After completing your first contribution task (instructions here) you need to Select a vignette from theproject boardthat is labeled ‘Easy’ or ‘Medium’, set to ‘Todo’ and are unassigned. More instructions you can find in the second contribution taskhere > 2. Currently all the issues on the project board under outreachy are assigned, the mentors will be releasing more vignette by the end of this week. > Hope it helps!
Monalika Patnaik (07:15:21): > @Paul Villafuerte@Andres WokatyKindly update the board with more vignettes as I have completed first contribution task and looking for the vignettes to work upon..Thankyou!
Kayla Okereke (09:01:58): > @Kayla Okereke has joined the channel
Madelyn Carlson (09:20:11): > Outreachy applicants: > > As a reminder, we are hosting the Sweave2Rmd-Outreachy office hour at 11-11:45AM ET today (Zoom link). This will be time where we explain the project and answer questions about the contribution tasks. If you’re unable to make it, no worries - we have another office hour session next week on Thursday, March 16, 10-10:45AM ET (and sessions each week for the 2 weeks after) and I will try my best to remember to record the session today and post it on this channel. Talk soon!
Namusisi Sharon (09:57:57): > @Namusisi Sharon has joined the channel
Shallie (10:34:27): > @Shallie has joined the channel
Job David (12:07:49): > @Job David has joined the channel
Job David (12:12:29): > Hello everyone, I am David from Nigeria, I look forward to contributing to the sweave2rmd project, please can anyone tell me how to get started on it??
Emmanuel Taiwo devbird007 (12:33:24) (in thread): > Welcome David, to get started, click this linkhttps://github.com/Bioconductor/sweave2rmd/issues/62to get taken to the issue where you will make your first contribution. It will require you to learn or refresh your knowledge on a few skills including R, RStudio and R Markdown, which are necessary for this project. Then you take a quiz which is also contained within the link. > > Next you can move over to this issuehttps://github.com/Bioconductor/sweave2rmd/issues/63where you can start picking sweave vignettes from the project board and making contributions. There’s instructions within to help you with everything you’ll need. > > Good luck - Attachment: #62 Sweave2Rmd Knowledge Check - Attachment: #63 Replace a Sweave Vignette With an R Markdown Vignette
Job David (12:34:11) (in thread): > Thanks@Emmanuel Taiwo devbird007
Treasure Okafor (15:11:23) (in thread): > Thank you so much@Monalika Patnaik..I would try to go on with these tips. I appreciate:pray:
OLADIRAN OLUWASHOLA (18:13:27): > @OLADIRAN OLUWASHOLA has joined the channel
2023-03-09
Sreeyusha Vajinapally (00:20:58): > @Sreeyusha Vajinapally has joined the channel
Ukpai Bishop (03:20:51): > @Ukpai Bishop has joined the channel
Ukpai Bishop (03:23:44): > Hi everyone, I am Bishop ukpai an outreachy intern. Please how do I start the contribution process
rohitsatyam102 (05:05:44) (in thread): > @Ukpai BishopMaybe this is how we get started.
Emmanuel Taiwo devbird007 (05:17:30) (in thread): > https://community-bioc.slack.com/archives/C03E7TWE2NT/p1678296804072499?thread_ts=1678295549.563899&cid=C03E7TWE2NT - Attachment: Attachment > Welcome David, to get started, click this link https://github.com/Bioconductor/sweave2rmd/issues/62 to get taken to the issue where you will make your first contribution. It will require you to learn or refresh your knowledge on a few skills including R, RStudio and R Markdown, which are necessary for this project. Then you take a quiz which is also contained within the link. > > Next you can move over to this issue https://github.com/Bioconductor/sweave2rmd/issues/63 where you can start picking sweave vignettes from the project board and making contributions. There’s instructions within to help you with everything you’ll need. > > Good luck
K Nodia (08:06:25): > @K Nodia has joined the channel
Ukpai Bishop (10:04:48) (in thread): > Thanks for the help
Job David (12:37:47) (in thread): > Hello good day@Madelyn CarlsonI hope this message finds you well. I wanted to follow up on the Zoom meeting we had during our last meeting. You mentioned that you would send the recorded link to join the meeting, but we haven’t received it yet. > > I would appreciate it if you could kindly send us the Zoom meeting link at your earliest convenience so that we can rewatch it. > > Thank you so much for your help and support.
Madelyn Carlson (12:38:46) (in thread): > hi@Job Davidthe recording link was posted in our#outreachychannel yesterday. Here is the link to the post/recording link:https://community-bioc.slack.com/archives/C037LN1HQDB/p1678309859829319 - Attachment: Attachment > For those of you who couldn’t make it to our first office hour for the Sweave2Rmd project, here is a recording of our discussion. Feel free to check it out and hear the questions we discussed. You can also tune in to our next office hour session on Thursday March 16, 10-10:45AM ET (Zoom link). After that the following sessions include: > • Wednesday, March 22, 11-11:45AM ET (Zoom link) > • Thursday, March 30, 10-10:45AM ET (Zoom link) > Please let us know if these times do not work for you - we are happy to adjust.
Madelyn Carlson (12:38:54) (in thread): > Hope it is useful!
Job David (12:39:24) (in thread): > Thanks:pray:for your response
Madelyn Carlson (12:39:28) (in thread): > absolutely!
Job David (12:39:58) (in thread): > Yeahit’llbe useful:smiling_face_with_3_hearts::smiling_face_with_3_hearts:
Sonali Kumari (14:52:12): > I am getting this Error anyone Knows the solution? - File (PNG): 2023-03-10 (1).png
{kind=link}
Marcel Ramos Pérez (14:58:00): > You should install the package withBiocManager::install("RNAseqData.HNRNPC.bam.chr14")
Sonali Kumari (15:01:00): > ohk
2023-03-10
Busayo Samuel (she/her) (03:56:00): > @Busayo Samuel (she/her) has joined the channel
Dennis Ndubi (04:28:13): > @Dennis Ndubi has joined the channel
Dennis Ndubi (04:30:08): > Greetings mentors and fellow applicants, Dennis Ndubi here, an Outreachy intern. I am interested in contributing to the project “Convert Sweave Vignettes to R Markdown”, thus I would like to connect with people who are already working on it for further guidance and help. Looking forward to building a great network and community. Thank you.@Andres Wokaty
Sonali Kumari (04:50:58) (in thread): > you can start with the first contribution task (instructions here) if interested in the Sweave2Rmd project. Once completed, you have the option of starting asecond (optional) contribution taskin which you claim a vignette from the project board and convert it. The first contribution task is designed as a ramp to the second task, which is why we ask that you work on them sequentially.#62 Sweave2Rmd Knowledge CheckBioconductor/sweave2rmd| Mar 6th | Added byGitHub#63 Replace a Sweave Vignette With an R Markdown VignetteBioconductor/sweave2rmd| Mar 6th | Added byGitHub
Job David (05:54:59) (in thread): > Hello@Emmanuel Taiwo devbird007do I need to clone my repo first before installing the devtools and biocManager ?@Andres Wokaty
Job David (06:33:42): > Please, how can I select a vignette and set the Status to In Progress and also how can assigned my name under it ??
Job David (06:39:58) (in thread): > @Sonali KumariPlease, how can I select a vignette and set the Status to In Progress and also how can assigned my name under it ??
Sonali Kumari (07:09:40): > you Just have to choose an unassigned vignette fromhttps://github.com/orgs/Bioconductor/projects/2/views/6and comment down herehttps://github.com/Bioconductor/sweave2rmd/issues/63that you want to work in that vignette . Then mentors will assign you that and update that in the project board - Attachment: #63 Replace a Sweave Vignette With an R Markdown Vignette
Sonali Kumari (07:11:59) (in thread): > you Just have to choose an unassigned vignette fromhttps://github.com/orgs/Bioconductor/projects/2/views/6and comment down herehttps://github.com/Bioconductor/sweave2rmd/issues/63that you want to work in that vignette . Then mentors will assign you that and update that in the project board (edited)#63 Replace a Sweave Vignette With an R Markdown VignetteBioconductor/sweave2rmd| Mar 6th | Added byGitHub
Andres Wokaty (07:38:05) (in thread): > Have you already donehttps://github.com/Bioconductor/sweave2rmd/issues/62? - Attachment: #62 Sweave2Rmd Knowledge Check > Welcome Outreachy applicants! > > Note: Multiple Outreachy applicants can work on this issue. > > Please choose this as your first contribution task to the Bioconductor Sweave2Rmd project as it will familiarize you with the R, R Markdown, GitHub processes, documentation, and the Sweave2Rmd conversion process (learn more about Bioconductor here). > > Sweave2Rmd Project > > Sweave2Rmd is a community project to convert Sweave vignettes to Rmd. Currently, there are approximately 900 vignettes .Rnw in software packages. These vignettes do not operate as effectively as .Rmd as they are older, combine latex with R, and compile to PDF whereas .Rmd vignettes compile to HTML, are BiocStyle compatible, and easier to read and maintain. Therefore, the aim of the Sweave2Rmd project is to convert these Sweave vignettes to .Rmd. > > Prep Work > > If you’re not familiar with R and R Markdown, please learn a little about them. The following links should be sufficient to get you started: R Primer - Programming basics or R Markdown. Then you may complete the following steps: > > • Install R > • Install RStudio > • In RStudio or through the terminal, install devtools and BiocManager:
> install.packages(c("devtools", "BiocManager")) > • Read through the Sweave2Rmd Contribute article, which explains step-by-step the process of converting a Sweave document to Rmd. The article will be important for completing this contribution task. > > Contribution Task > > After reading the Sweave2Rmd Contribute article, you may complete the 2023 Outreachy Sweave2Rmd Contribution Familiarity Quiz. The quiz contains questions about the process of converting a Sweave vignette to Rmd. It is an opportunity to both test your familiarity with the Contribute article and learn how to convert a vignette if you choose to complete the second (optional) contribution task. We expect the quiz to take up to 3 hours to complete. If you have any questions about the quiz, feel free to post on the Sweave2Rmd Outreachy Slack channel. To join the Slack channel, view https://slack.bioconductor.org/ then search for the channel #outreachy. > > Remember to record your contributions in Outreachy at https://www.outreachy.org/outreachy-may-2023-internship-cohort/communities/bioconductor/convert-sweave-vignettes-to-r-markdown/contributions/
sophy (08:52:10): > @sophy has joined the channel
Emmanuel Taiwo devbird007 (08:59:08) (in thread): > @rohitsatyam102
Emmanuel Taiwo devbird007 (09:00:41) (in thread): > You need to complete the first contribution where you will be required to learn a few concepts relating to the project, then take the familiarity quiz
rohitsatyam102 (09:01:52) (in thread): > Yes I completed the quiz yesterday
Emmanuel Taiwo devbird007 (09:03:47) (in thread): > Good job, now you can move on to the second part of the contribution phase where you select an unassigned vignette and comment under the issue that you would like to work on it.
Madelyn Carlson (11:16:31) (in thread): > Thank you@Sonali Kumari! Please let us know if you have additional questions once you’ve reviewed the contribution task guidance,@Dennis Ndubi. Happy to have you here
Ukpai Bishop (11:49:24): > Please is there a deadline for the contribution
Madelyn Carlson (11:49:52) (in thread): > Great question, no there is no deadline for the first Sweave2Rmd contribution task as long as it is done within the contribution period
Ukpai Bishop (11:50:47) (in thread): > Thank you very much Madelyn
Dennis Ndubi (12:02:35): > hello everyone. Once i have been assigned to a certain vignette to make a contribution to, how would I go about it@Andres Wokaty.
Andres Wokaty (12:05:15) (in thread): > Hi Dennis, if you haven’t done the Knowl edge quiz athttps://github.com/Bioconductor/sweave2rmd/issues/62, I recommend starting there first because it will help familiarize you with the project. Then dohttps://github.com/Bioconductor/sweave2rmd/issues/63. Please read all of the issue info and follow the links. It will tell you how to do the contribution taks. - Attachment: #62 Sweave2Rmd Knowledge Check - Attachment: #63 Replace a Sweave Vignette With an R Markdown Vignette
Dennis Ndubi (12:08:30) (in thread): > thank you… i will keep in touch incase of any challenge
Ukpai Bishop (15:03:20): > Hi, please how do I know an issue that is open
Madelyn Carlson (15:04:17) (in thread): > to clarify, do you mean available vignettes? i.e., how to tell whether a vignette is unclaimed ?
Ukpai Bishop (15:04:40) (in thread): > Yes
Madelyn Carlson (15:06:36) (in thread): > You can check out ourproject board, review the vignettes that do not have an “assigned” person and comment on theissues pageto say which one you’d like to convert. We will upload your name on the “assigned” column once you’ve commented on issues pg - Attachment: #63 Replace a Sweave Vignette With an R Markdown Vignette
khadijah (15:10:54): > @khadijah has joined the channel
Dennis Ndubi (15:32:33) (in thread): > hello…i need some help here… i have accessed the Gostats package at the BiocViews landing page. my question is, should i clone the package repo into a local repo where i should convert the vignette to Rmd and the upload my local repo or what should i do.. kindly assist@Andres Wokaty - File (PNG): image.png
{kind=link}
Dennis Ndubi (15:39:56) (in thread): > hello…i need some help here… i have accessed the Gostats package at the BiocViews landing page. my question is, should i clone the package repo into a local repo where i should convert the vignette to Rmd and the upload my local repo or what should i do.. kindly assist@Andres Wokaty.. - File (PNG): image.png
{kind=link}
Andres Wokaty (15:41:52) (in thread): > It’s upto you if you prefer to clone it or just copy and paste it into a file. For that question, you are only submitting the DESCRIPTION file. You should imagine that you’re converting one of its vignettes in this scenario.
Dennis Ndubi (15:44:23) (in thread): > okay.. thank you
2023-03-11
Muluh (02:04:23): > @Muluh has joined the channel
Muluh (02:04:55): > Hello, I am Daena. I am excited about working on this project “Convert Sweave Vignettes to R Markdown”. My pronouns are she/her.
MaryJones (08:49:58): > Hello:
MaryJones (08:53:43): > please can someone kindly explain how to fix the pandoc error, here is a screenshot. I followed all instructions but dont know if am still missing something, I am a novice but a fast learner too any suggestion will be appreciated , thank you:pray: - File (PNG): pandoc issue in Rstudio at 3.16.38 PM.png
{kind=link}
Sonali Kumari (09:37:15): > I am getting this error while running tar ztf GenomicFiles.tar.gz | grep ‘doc/GenomicFiles’ command in R . Please help me if anyone knows the answer - File (PNG): 2023-03-11 (4).png
{kind=link}
Emmanuel Taiwo devbird007 (10:01:55) (in thread): > Hi Daena! Welcome to the sweave2rmd slack channel. A good place to start is to access the first issuehttps://github.com/Bioconductor/sweave2rmd/issues/62. Go through it and take the quiz attached. This will prepare you for the next phase. > > Once you’re done, you can access the next issue here:https://github.com/Bioconductor/sweave2rmd/issues/63It contains everything you’ll need to know to begin contributing and converting sweave vignettes to Rmd. - Attachment: #62 Sweave2Rmd Knowledge Check - Attachment: #63 Replace a Sweave Vignette With an R Markdown Vignette
Andres Wokaty (10:03:14) (in thread): > You need to run the tar command in a terminal, not the R console.
Emmanuel Taiwo devbird007 (10:07:40) (in thread): > Hey Mary, the error indicates that there isn’t ChromBond.Rnw file in your current working directory. You need to switch to where the file is located and then run the command
Sonali Kumari (10:08:00) (in thread): > ohk thanku so much
MaryJones (10:12:38) (in thread): > ok, let me check thanks
MaryJones (10:18:11) (in thread): > @Emmanuel Taiwo devbird007I am in the directory, I used getwd() to check it is the same directory, but do I need to set the working directory to the find the vignette folder ?
Sonali Kumari (10:26:01) (in thread): > I am again getting this error - File (PNG): 2023-03-11 (5).png
{kind=link}
Andres Wokaty (10:26:58) (in thread): > You are missingtarat the beginning of the statement
Sonali Kumari (10:51:09) (in thread): - File (PNG): 2023-03-11 (6).png
{kind=link}
Sonali Kumari (10:51:39) (in thread): > I am using windows.
Sonali Kumari (10:52:19) (in thread): > trying in project terminal as well as in command prompt.
Sonali Kumari (10:55:02): > getting this error in terminal , I am using windows try running code in project terminal as well as in command prompt. If anyone anyone can help. - File (PNG): 2023-03-11 (6).png
{kind=link}
MaryJones (11:56:09) (in thread): > Ok, thank you.. yeap just check it now and its installed . I guess I will need to set my path. Thanks alot, I appreciate:pray:
Emmanuel Taiwo devbird007 (12:21:41) (in thread): > Hi Sonali, a couple of things you should check are: > 1. You are directly outside the cloned repository on your system > 2. You have to run it just as it is in the Contributing Article: > >tar ztf GenomicFiles.tar.gz | grep doc/GenomicFiles
Emmanuel Taiwo devbird007 (12:24:27) (in thread): > That’s good to hear, glad it worked in the end.
Janani Ravi (12:55:19): > @Janani Ravi has joined the channel
Andres Wokaty (17:16:39) (in thread): > I don’t see the tar ball in your directory. The tar ball is created from runningR CMD build GenomicFileswhereGenomicFilesis your package’s directory.
2023-03-12
Sonali Kumari (00:54:18) (in thread): > thanks@Emmanuel Taiwo devbird007
Sonali Kumari (06:10:20) (in thread): > @Dennis Ndubiyou just have to upload the converted .Rmd file with the YAML block in top .
Aliyu Atiku Mustapha (07:05:07): > @Aliyu Atiku Mustapha has joined the channel
Sonali Kumari (10:42:39) (in thread): - File (PNG): 2023-03-12 (4).png
{kind=link}
Sonali Kumari (10:43:06) (in thread): > all done@Andres WokatyThank you so much for your Support.
Dennis Ndubi (13:53:26) (in thread): > okay.. thank you for the assistance
Dennis Ndubi (15:43:32): > hello everyone. i am finding difficulties to complete the knowledge check. i have gone through the contribution article, and selected the one vignette from theSweave2Rmd project boardand i am stuck from there…any help will be appreciated@Andres Wokaty
Sonali Kumari (15:58:35) (in thread): > hello@Dennis Ndubiyou just have to follow the steps fromhttps://github.com/Bioconductor/sweave2rmd/issues/62 - Attachment: #62 Sweave2Rmd Knowledge Check > Welcome Outreachy applicants! > > Note: Multiple Outreachy applicants can work on this issue. > > Please choose this as your first contribution task to the Bioconductor Sweave2Rmd project as it will familiarize you with the R, R Markdown, GitHub processes, documentation, and the Sweave2Rmd conversion process (learn more about Bioconductor here). > > Sweave2Rmd Project > > Sweave2Rmd is a community project to convert Sweave vignettes to Rmd. Currently, there are approximately 900 vignettes .Rnw in software packages. These vignettes do not operate as effectively as .Rmd as they are older, combine latex with R, and compile to PDF whereas .Rmd vignettes compile to HTML, are BiocStyle compatible, and easier to read and maintain. Therefore, the aim of the Sweave2Rmd project is to convert these Sweave vignettes to .Rmd. > > Prep Work > > If you’re not familiar with R and R Markdown, please learn a little about them. The following links should be sufficient to get you started: R Primer - Programming basics or R Markdown. Then you may complete the following steps: > > • Install R > • Install RStudio > • In RStudio or through the terminal, install devtools and BiocManager:
> install.packages(c("devtools", "BiocManager")) > • Read through the Sweave2Rmd Contribute article, which explains step-by-step the process of converting a Sweave document to Rmd. The article will be important for completing this contribution task. > > Contribution Task > > After reading the Sweave2Rmd Contribute article, you may complete the 2023 Outreachy Sweave2Rmd Contribution Familiarity Quiz. The quiz contains questions about the process of converting a Sweave vignette to Rmd. It is an opportunity to both test your familiarity with the Contribute article and learn how to convert a vignette if you choose to complete the second (optional) contribution task. We expect the quiz to take up to 3 hours to complete. If you have any questions about the quiz, feel free to post on the Sweave2Rmd Outreachy Slack channel. To join the Slack channel, view https://slack.bioconductor.org/ then search for the channel #outreachy. > > Remember to record your contributions in Outreachy at https://www.outreachy.org/outreachy-may-2023-internship-cohort/communities/bioconductor/convert-sweave-vignettes-to-r-markdown/contributions/
Emmanuel Taiwo devbird007 (16:20:52) (in thread): > Hi Dennis, number 2 in the “Create the R Markdown” section of theSweave2Rmd Contribute articlesays, after you select a vignette, you should search for the package onBiocViews, then continue with the following steps to understand how you should make your contribution. > > Does this solve your problem?
Dennis Ndubi (16:37:52) (in thread): > i have found the package on the BiocViews, and accessed the PDF of the vignette, how do IFork the repositoryi should work on work on
Andres Wokaty (16:41:42) (in thread): > For the knowledge quiz, you don’t have to fork the repo. You are just pretending you that you are working on it.
Dennis Ndubi (16:47:12) (in thread): > okay, I have got it, that means I can create any random file to showcase i have understood the contents of the contribution article and how to convert Rnw to a Rmd.. thanks
Emmanuel Taiwo devbird007 (17:05:18) (in thread): > Whatever file you create, it would have to be based on a vignette you have chosen from the project board.
2023-03-13
Dennis Ndubi (08:36:50): > hello.. could someone kindly tell me if i am in the right direction in answering question one of the knowledge check.. i have attached the file..@Andres Wokaty..any instructions and assistance is appreciated - File (HTML): YAML.html - File (Plain Text): YAML.Rmd
Parul Chaddha (08:56:40): > @Parul Chaddha has joined the channel
Reece (09:02:05) (in thread): > @Dennis Ndubithis looks like a demo template and yes that’s the right path, proceed with the answering
Dennis Ndubi (10:15:48) (in thread): > ok.. thank you
Andres Wokaty (10:26:29) (in thread): > The .Rmd file looks like the better option. You just need the yaml part for the question, which is the part that is between the---
Reece (11:21:24): > @Andres Wokaty@Paul Villafuerteam running into this error can’t locate package GeneMeta, anyone with a walk around - File (PNG): errorr.png - File (PNG): errorr2.png
{kind=link}
{kind=link}
Andres Wokaty (11:23:59) (in thread): > I recommend usingBiocManager::install()to installGeneMeta.BiocManageris the recommended tool for installing Bioconductor packages. It will attempt to install the correct version for your version of R and include any dependencies.
Reece (11:28:06) (in thread): > @Andres Wokatyworks like magic:sparkles:Thank you
Dennis Ndubi (11:29:29) (in thread): > thank you@Andres Wokatymy question is solved.
Reece (11:30:19) (in thread): > For some reason, Rstudio first loads with a version less than 4.3 the recommended for this project why?
Andres Wokaty (11:31:41) (in thread): > So R and Bioconductor have a “release” and a “devel” branch/model at all times. Release is the stable version whereas devel is the future release coming up next month, which can still have bugs.
Dennis Ndubi (12:06:50): > hello again.. is this the right way to answer question 2 of the knowledge check(-attaching file)…Any corrections and instructions to achieve the right solution will be appreciated@Andres Wokaty - File (Plain Text): Description.Rmd
Andres Wokaty (12:12:29) (in thread): > You are on the right track. I would fix theAuthors@Rsection (names, the A at the beginning, name of the vignette) and put a space beforeknitr.
Dennis Ndubi (12:27:55) (in thread): > Thumbs up for the help, it was resourceful.
Dennis Ndubi (15:07:53): > Hello, can someone kindly help me understand How do you link to Bioconductor packages? am having some difficulties there@Andres Wokaty
Sonali Kumari (15:48:13) (in thread): > you can refer this to understand the concepthttps://bioconductor.org/packages/release/bioc/vignettes/BiocStyle/inst/doc/AuthoringRmdVignettes.html
Dennis Ndubi (16:26:38) (in thread): > i have still not understood how to go about iit
Andres Wokaty (16:29:20) (in thread): > I think it would be better to ask specific questions about what you don’t understand. The document@Sonali Kumarishared has the answer.
Taofeekat Olorunkosebi (20:09:57): > @Taofeekat Olorunkosebi has joined the channel
Taofeekat Olorunkosebi (20:20:39): > Hello everyone, > I’m Taofeekat Olorunkosebi (She/her) from Nigeria, I have a background in software development and I’d like to work on ‘Convert Sweave Vignettes to R Markdown’ project. I’m looking forward to networking with like minds and learn more from the community while making valuable contributions. > > Thank you.
2023-03-14
Busayo Samuel (she/her) (00:41:41) (in thread): > Welcome Taofeekat
Sonali Kumari (02:06:36) (in thread): > Hi@Andres Wokatyi found out that for wrapping long lines we need to highlight the long paragraph and run `Code > Reflow Comment but i was anable to do it correctly please guide me how to do that correctly. - File (PNG): 2023-03-14.png
{kind=link}
Andres Wokaty (02:08:40) (in thread): > At the top left menu, select Reflow Comment from the Code menu
Sonali Kumari (02:09:08) (in thread): > ohk.
Sonali Kumari (02:09:49) (in thread): > thanku so much@Andres Wokatyit worked .
Emmanuel Taiwo devbird007 (04:01:52) (in thread): > Hello there Taofeekat, to get started, click this linkhereto get taken to the issue where you will make your first contribution. It will require you to learn or refresh your knowledge on a few skills including R, RStudio and R Markdown, which are necessary for this project. Then you take the quiz which is also contained within the issue. > > Next you can move over to this issueherewhere you can start picking sweave vignettes from the project board and making contributions. There’s instructions within to help you with everything you’ll need. > > Good luck.
Taofeekat Olorunkosebi (06:08:23) (in thread): > Thank you so much
Dennis Ndubi (07:14:39): > Must i fork the repository or i can just clone it into the local repo using the given clone link directly? Kindly give necessary directions@Andres Wokaty - File (PNG): image.png
{kind=link}
Aliyu Atiku Mustapha (08:24:03) (in thread): > You must Fork before you Clone
Andres Wokaty (08:54:15) (in thread): > What are you working on?
Dennis Ndubi (09:07:49) (in thread): > I want to complete the second contributionhttps://github.com/Bioconductor/sweave2rmd/issues/63and the vignette assigned to me is GOstats/vignettes/GOstatsHyperG.Rnw but i am somehow stuck - Attachment: #63 Replace a Sweave Vignette With an R Markdown Vignette > Hello Outreachy applicants! > > Note: This task can be completed by multiple Outreachy applicants. Please choose this as your second (optional) contribution task to the Bioconductor Sweave2Rmd project as it builds upon the familiarity quiz in the Sweave2Rmd first contribution. > > Prep Work > > • Sweave2Rmd Knowledge Check > • Please read our contribution document, which gives a good overview of the project plus the general workflow. In particular, you will be performing the tasks from <https://bioconductor.github.io/sweave2rmd/articles/contribute.html#create-the-r-markdown–rmd-vignette-and-remove-the-sweave-vignette–rnw|Create the R Markdown (.Rmd) vignette and Remove the Sweave vignette (.Rnw)>. > • Install pandoc > > Contribution Task > > Make a pull request replacing a Sweave vignette with an R Markdown vignette. > > 1. Select a vignette from our project board that is labeled ‘Easy’ or ‘Medium’, set to ‘Todo’ and are unassigned. (As a FYI, although there is no maximum # of vignettes you’re allowed to convert, project leads must review everyone’s first PR before we are able to assess any additional vignettes from anyone contributing to the project). > 2. Write a comment with the path of the vignette you’d like to do–e.g., BiocParallel/vignettes/Introduction_To_BiocParallel.Rnw. We will write your GitHub username as ‘Assigned’ and set ‘Last Modified’ to today. > 3. Perform the steps in <https://bioconductor.github.io/sweave2rmd/articles/contribute.html#create-the-r-markdown–rmd-vignette-and-remove-the-sweave-vignette–rnw|Create the R Markdown (.Rmd) vignette and Remove the Sweave vignette (.Rnw)>. Note: You do not need to contact the maintainers to work on this set of vignettes as they have either been previously contacted OR they are maintained by the Bioconductor Core Team. > 4. Make sure that you request a review by writing @Bioconductor/sweave2rmd in a comment so that we can review your pull request. For example, @Bioconductor/sweave2rmd please review my contribution for Outreachy. > 5. Make a comment in this issue with a link to your PR. > 6. If you receive any feedback requesting changes, please address them. > > Tip: You can find the PDF you’ll be replacing by visiting the landing page for the package under Documents. You can look up the package at <https://bioconductor.org/packages/devel/BiocViews.html#___Software|Bioconductor Software Packages>.
> Tip: You can read how we <https://bioconductor.github.io/sweave2rmd/articles/contribute.html#review-a-pull-request|review PRs> in the contribution document. Use the checklist to assess if your PR will pass review. > > If your pull request is good, it will be merged into Bioconductor! > > Remember to record your contributions in Outreachy at https://www.outreachy.org/outreachy-may-2023-internship-cohort/communities/bioconductor/convert-sweave-vignettes-to-r-markdown/contributions/.
Andres Wokaty (09:09:50) (in thread): > Please read the instructions and visit all the links because they contain some of the answers you are looking for.~Before you work this contribution, you should request a vignette to work on that way no two applicants work on the same vignette.~I see you are Dbn-tech.
Andres Wokaty (09:14:09) (in thread): > I see the issue now.
Andres Wokaty (09:15:32) (in thread): > Don’t clone using the links on the package’s landing page forgit.bioconductor.org:packages/GOstats. These links go to Bioconductor’s private git whereas we want to work fromgithub.com. So you should start fromgithub.com/Bioconductor/GOstats
Andres Wokaty (09:16:14) (in thread): > We do this so that we can use github’s pull request functionality to review changes.
Andres Wokaty (09:16:25) (in thread): > Does that make sense?
Dennis Ndubi (09:16:41) (in thread): > yes.. it makes sense now
Dennis Ndubi (09:18:37) (in thread): > my issue is solved and I can now continue. Thank you so much
Elisheba Asiimwe Joanita (10:00:43): > @Elisheba Asiimwe Joanita has joined the channel
Sonali Kumari (16:03:52): > @Andres Wokatyerror message occur while doing this change in the .Rmd file i guess i am doing some mistake please correct me . - File (PNG): 2023-03-15 (2).png
{kind=link}
Sonali Kumari (16:04:59) (in thread): > i think we should change the label as the error msg says.
Sonali Kumari (16:05:15) (in thread): > please correct me
Andres Wokaty (16:14:30) (in thread): > I see. The message says it’s a duplicate label, so maybe you need to change it from ‘#fig1’ to ‘#figure1’, for example.
Andres Wokaty (16:14:53) (in thread): > I also noticed that you’re putting the anchor right next to the number, but the anchor should be right above the figure.
Sonali Kumari (16:16:41) (in thread): > ohk I will do the needful changes.
Sonali Kumari (16:29:41) (in thread): - File (PNG): 2023-03-15 (4).png
{kind=link}
Sonali Kumari (16:31:40) (in thread): > again same error
Sonali Kumari (16:42:41) (in thread): > I have also tried with different label names but it is showing same error
Andres Wokaty (17:20:17) (in thread): > You should change the anchor too. Code blocks must have unique names. It’s not clear if it is this new code or another piece of code. I would temporarily remove it and see if you can knit. If you can knit, you know this is the problem code. I’ll try to find a reference to another .Rmd that does this but I probably won’t be able to get to you until tomorrow
Sonali Kumari (17:22:16) (in thread): > i am trying something if it works i will let you know
Sonali Kumari (17:24:48) (in thread): > no issues I will try to find a solution myself if it works I will let you know if not then you can help me tomorrow anytime.
Sonali Kumari (17:33:34) (in thread): > After going through the other code blocks I found out that there is a code block which is actually a problem:sweat:. but the good part is that the problem is solved. Thank you@Andres Wokaty.
2023-03-15
Dennis Ndubi (00:06:29): > Hello, I keep encountering this error while converting individual code chunks. Though once I remove the results=hide option, knitr works and renders. Should I keep removing the chunk option to avoid the error@Andres Wokaty - File (PNG): image.png
{kind=link}
MaryJones (02:41:18): > Hello everyone:
MaryJones (02:42:38): > Please what could probably be causing this error ?: Error at “ChromBand.Rnw” (line 747, column 2): > unexpected end of input. - File (PNG): new pandoc issue_file not converting.png
{kind=link}
MaryJones (02:45:03): > I tried everything I know nothing seems to be working, I Monterey Os and the highest R update is 4.2.2, please any ideas , I am literally pulling my hairs out right now, am so confused:thinking_face:. Please kindly help , thank you all,:pray:
Dennis Ndubi (02:55:20) (in thread): > thank you.. lem try it out
Sonali Kumari (03:00:27) (in thread): > @Dennis NdubiI have changed the the msg bit please see actually when we want only results to be displayed and want to hide rest of the messages during execution of code we use message=false and if we want non of them to be displayed in the html document then we use include =false
Sonali Kumari (03:01:40) (in thread): > you should try with message =false
khadijah (03:09:46) (in thread): > Can you show me line 747
Levi Waldron (03:17:04) (in thread): > Mary is this the original Rnw file or after modifications?
MaryJones (03:20:11) (in thread): > @khadijahHello, there is nothing in line 747
MaryJones (03:20:50) (in thread): > @Levi WaldronHello, yes sir , that is the Raw file
MaryJones (03:22:32): > Ok So I am able to get an Rmd file generated by using this solution I got from pandoc site documentation:pandoc -o file.Rmd file.Rnw
MaryJones (03:27:25): > Now this is the new output of the .Rmd file and I am getting a new error I dont understand, here is the screenshot and short video file so you may understand how to guide me and advice on fixing it .. Thank you everyone and all the experts here, I appreciate all your help,:pray: - File (PNG): pandoc new error.png - File (MPEG 4 Video): new pandoc Rmd output.mp4
{kind=link}
Dennis Ndubi (03:28:21) (in thread): > and remember the pandoc has issues of converting code chunks therefore i remind you to change them manually into markdown until you get the perfect html output after knitr
Levi Waldron (03:32:45) (in thread): > That suggests to me that it is not a problem in the file, but some thing with how are you are trying to knit it?
khadijah (03:41:55) (in thread): > I can jump on a call with you if you don’t mind@MaryJoneswe can check it together
Dennis Ndubi (04:18:08): > I have noticed that after converting my Rnw to Rmd, some code lines are missing in the code chunk (as well as the html output) as compared to the pdf documentation of the vignette. Should i add the code lines to the code chunk manually too? - File (PNG): image.png
{kind=link}
Dennis Ndubi (04:18:57) (in thread): > @Andres Wokaty@Sonali Kumari
MaryJones (04:20:26) (in thread): > @Dennis NdubiHi, Ok, I see coz I was wondering may if I can preview the final output before adding to git. thanks a lot will do just that
MaryJones (04:22:15) (in thread): > @khadijahNot a problem, did you see the output ?, we can have a call when you free, just let me know I will call you. Thanks alot, really appreciate,:pray:
Levi Waldron (04:28:36) (in thread): > That looks like the original author “cheated” by making static comments look like code output?Definitely not best practice in vignettes unless it is absolutely unavoidable for some reason…
Levi Waldron (04:29:35) (in thread): > Can you turn them into real code that runs from a real knitr code chunk?
Levi Waldron (04:31:43) (in thread): > But your output looks like what should be produced by your Rmd
Sonali Kumari (04:33:21) (in thread): > @Dennis Ndubican you please show the picture of original .Rnw file i want the picture of that part which is missing in .Rmd file then I think maybe I can help you.
Dennis Ndubi (05:29:02) (in thread): > let me check where i have made a mistake then i will consult back.. thank you
MaryJones (07:54:40) (in thread): > @Levi WaldronAre you responding to me sir ?
Dennis Ndubi (07:56:21): > I am having challenges converting this code chunk since i have been trying and it gives an error. Can someone kindly advise on what to do@Andres Wokaty@Levi Waldron - File (PNG): image.png
{kind=link}
MaryJones (07:56:40) (in thread): > @Andres Wokaty@Sonali Kumari@Levi Waldronplease what must I do now. At the moment I am using html5 syntax to modify the file.
MaryJones (07:56:52) (in thread): > please advice , thank you all
Andres Wokaty (08:24:19) (in thread): > Looking at the screenshot, ‘hide’ is a string, so you need to put it in quotes:results="hide".TRUEis a logical value, so it doesn’t need the quotes. Also, on line 100, this code block should start withrso it should read > ```
Sonali Kumari (08:25:35) (in thread): > thanks@Andres Wokatywell noted.
Dennis Ndubi (08:57:16) (in thread): > i have solved the problem. i am good to go
Andres Wokaty (08:57:42) (in thread): > I would also refer to the existing PDF to help guide you in the process:https://bioconductor.org/packages/3.17/bioc/vignettes/GOstats/inst/doc/GOstatsHyperG.pdf
Dennis Ndubi (08:59:40) (in thread): > Thank you
Andres Wokaty (09:13:05) (in thread): > I don’t think that the command will give you R markdown content. We should try to use the original which attempts to convert the latex to markdown.
Rishi Singh (09:13:08): > @Rishi Singh has joined the channel
Andres Wokaty (09:51:36) (in thread): > It looks like the file has 2 issues when trying to convert: > 1. problem with underscores > 2. ‘end of file’ > 3. problem with $ > The end of file issue is the hardest to deal with since you have to hunt for it, but I’ll tell you that it’s related to thechr12ideogram. The work around is usually to remove this section entirely, convert, then add it manually but in the R Markdown format. > > Regarding 1 and 3, you could substitute some value before the conversion then replace them after the conversion. > > I recommend also usinggit diffto help you remember what’s changed.
Dennis Ndubi (11:59:53): > hello, ..I would like a review on my prhttps://github.com/Bioconductor/GOstats/pull/2and give the necessary directions. Thank you@Andres Wokaty - Attachment: #2 G ostats hyper g rmd
Dennis Ndubi (12:08:33): > Attached are the files@Andres Wokaty@Levi Waldron..Kindly make review and advise. Thanks - File (Binary): GOstatsHyperG.Rmd - File (HTML): GOstatsHyperG.html
Andres Wokaty (12:09:56): > Please be patient regarding reviews. I will assign a reviewer soon, but I have a lot of meetings today. > > If anyone is waiting on a review, please use the checklist in your PR to review your work. Don’t wait for the reviewer to say fix this. Everyone should be using the checklist to identify issues to fix. The more you fix your PR the easier it will be for us review.
Dennis Ndubi (12:33:11): > got it..
Paul Villafuerte (15:22:17) (in thread): > @Dennis NdubiI’ve completed most of your review. I will complete this evening.
Dennis Ndubi (15:24:59) (in thread): > Thank you@Paul Villafuertewill be waiting for feedback. Nice time
Paul Villafuerte (15:26:04) (in thread): > You should see some comments that you can start working on
Dennis Ndubi (15:31:33) (in thread): > okay
2023-03-16
Emmanuel Taiwo devbird007 (03:12:04): > Good day everyone :-)
Emmanuel Taiwo devbird007 (03:54:17) (in thread): > Hi Dennis, you are to attach the PDF file and the HTML output to your pull request. Not send it to the slack channel.
Emmanuel Taiwo devbird007 (03:58:05) (in thread): > Hi MaryJones, have you been able to make progress on these problems yet?
Madelyn Carlson (10:21:06): > Thank you to folks on our office hours today pointing out that our office hours overlap with BugSigDB office hours. In case y’all are interested in attending both sessions, we’re switching the Sweave2Rmd office hour in 2 weeks from Thursday, March 30 to Wednesday, March 29 at 10-10:45AM ET. So here is the schedule for the next two weeks. > * Wednesday, March 22, 11-11:45AM ET (Zoom link) > * Wednesday, March 29, 10-10:45AM ET (Zoom link) > Hope to see you there
Aliyu Atiku Mustapha (11:16:08): > @Andres WokatyHi, I have a different .Rnw file which hasn’t been converted to .Rmd file and it is stopping my “R CMD build” from running its full course. > Question is, do I remove the file, run the commands for build and “tar ztf…” then put the file back?
Andres Wokaty (11:18:08) (in thread): > You shouldn’t have to remove other .Rnw files. The purpose of doingR CMD buildis to simulate what the build system will do to the package. Can you share the error you are getting?
Aliyu Atiku Mustapha (11:24:07) (in thread): > @Andres WokatyAt the vignettes folder there is a “graphAttributes.Rnw” file, but my work is on “graph” which I have removed the .Rnw file already. Which the second screenshot illustrates. The first illustrates the error I get running “R CMD build ….” - File (PNG): image.png - File (PNG): image.png
{kind=link}
{kind=link}
Andres Wokaty (11:58:06) (in thread): > I’m not sure what is the exact fix to your error. On windows machines, we usually use Miktex to help build pdfs:https://miktex.org/. Miktex appears to have grfext.sty.
Aliyu Atiku Mustapha (12:30:39) (in thread): > Ok, thank you. I will give Miktex a try
Dennis Ndubi (13:24:15): > Hello, could someone direct me how to achieve the indentation format in the original vignette(Sample Frame one)..my output after knitr(Frame two).Thank you.@Andres Wokaty - File (PNG): image.png - File (PNG): image.png
{kind=link}
{kind=link}
Aliyu Atiku Mustapha (13:36:09) (in thread): > Success!:raised_hands:So after installing Miktex, still encountered some errors mainly due to write permission. So I had to enable folder control to RStudio through the Windows Defender App. After that, it ran successfully. Also thecheck command as illustrated in the screenshot below. - File (PNG): image.png
{kind=link}
Paul Villafuerte (13:54:11) (in thread): > Take a look athttps://bookdown.org/yihui/rmarkdown-cookbook/indent-text.html. It does require some fiddling. - Attachment (bookdown.org): 5.2 Indent text | R Markdown Cookbook > This book showcases short, practical examples of lesser-known tips and tricks to helps users get the most out of these tools. After reading this book, you will understand how R Markdown documents are transformed from plain text and how you may customize nearly every step of this processing. For example, you will learn how to dynamically create content from R code, reference code in other documents or chunks, control the formatting with customer templates, fine-tune how your code is processed, and incorporate multiple languages into your analysis.
Dennis Ndubi (13:56:02) (in thread): > Thank you
Emmanuel Taiwo devbird007 (16:37:14) (in thread): > Looking forward to it
USLACKBOT (17:08:33): > This message was deleted.
Sonali Kumari (17:08:55) (in thread): > https://github.com/Bioconductor/GenomicFiles/pull/8#discussion_r1139335988
Andres Wokaty (17:10:20) (in thread): > So I think it should be placed right above the image so that when you click on it, the image is at the top.
Sonali Kumari (17:12:51) (in thread): > Ohk then I will do that Thanks for clarification
Sonali Kumari (17:13:19) (in thread): > What about this ?https://github.com/Bioconductor/GenomicFiles/pull/8#discussion_r1139347756
Andres Wokaty (17:16:49) (in thread): > I just responded to that one in the comments. I am finishing up the review.
Sonali Kumari (17:18:16) (in thread): > this is giving error
Andres Wokaty (17:18:46) (in thread): > What are you trying to do? that is html so it won’t be able to render that in the R console.
Andres Wokaty (17:19:06) (in thread): > knitr will be able to handle it, though.
Andres Wokaty (17:19:17) (in thread): > AH, I see
Andres Wokaty (17:19:38) (in thread): > please put it outside the code block, on line 155
Andres Wokaty (17:19:50) (in thread): > (Sorry, I was initially looking at the console)
Sonali Kumari (17:21:28) (in thread): > Ohk .
Sonali Kumari (17:23:30) (in thread): > @Andres Wokatyi still thinks that the initial position of these anchors are fine. because they are working fine with their functionality
Sonali Kumari (17:26:54) (in thread): > Please correct me.
Sonali Kumari (17:28:09) (in thread): > you can see herehttps://rpubs.com/Sonali8434/1014072
Andres Wokaty (17:29:03) (in thread): > Yes, they work; however, I am taking issue with the location. It should be right above the image.
Sonali Kumari (17:30:09) (in thread): > Ohk …. Thanks for clarification , I will do the changes right away.
2023-03-17
Busayo Samuel (she/her) (12:41:08): > Hi@Andres Wokatythanks your input on the Rsamtools vignette issue I was having. I just figured out I was supposed to run R CMD check on the tar ball result gotten from R CMD build, so everything looks good so far.:thumbsup:Cc@Madelyn Carlson
Dennis Ndubi (14:03:25): > hello, how does one R CMD build on RStudio@Andres Wokaty
Madelyn Carlson (14:05:34) (in thread): > To run R CMD build, you can type “R CMD build [name of package]” in your R terminal. We go through this step by step in the office hour recording from yesterday, if you’d like to check it out. Here is the link:https://us02web.zoom.us/rec/share/UNBJXgszTprwr293bu-6XmbmebSSaorMVjfn_LnFpuoftNqemQ1l5MFHneRhM-PA.UXF3hrpbM6df5vva
Madelyn Carlson (14:05:51) (in thread): > Thank you for the heads up!
Dennis Ndubi (14:06:36) (in thread): > thank you
Madelyn Carlson (14:06:46) (in thread): > Np!
2023-03-18
Dennis Ndubi (07:45:34): > Hello, while doing the R CMD build I am encountering this, Any directions on how to solve it@Andres Wokaty@Madelyn Carlson - File (PNG): image.png - File (PNG): image.png - File (PNG): image.png - File (PNG): image.png
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Busayo Samuel (she/her) (08:44:43) (in thread): > Have you tried installing multtest and the .sty package?
Dennis Ndubi (09:00:26) (in thread): > no i have not
Busayo Samuel (she/her) (09:02:10) (in thread): > https://github.com/jankapunkt/latexcv/wiki/!-LaTeX-Error:-File–filename-.sty-not-foundHere’s a link to help with the .sty issue
Dennis Ndubi (09:04:06) (in thread): > lem check it out
Dennis Ndubi (09:52:50) (in thread): > My problem is not solved since i am not using Linux I cant run the commands on the link you provided@Busayo Samuel (she/her)
Busayo Samuel (she/her) (10:11:10) (in thread): > From the error message, you’re having issues because your folder has .Rnw files and it’s having difficulties building the pdf for them. Other people have had this issue, you can go through this conversation to get more insight. I think you need Miktexhttps://community-bioc.slack.com/archives/C03E7TWE2NT/p1678979768272669?thread_ts=1678979768.272669&cid=C03E7TWE2NT - Attachment: Attachment > @Andres Wokaty Hi, I have a different .Rnw file which hasn’t been converted to .Rmd file and it is stopping my “R CMD build” from running its full course. > Question is, do I remove the file, run the commands for build and “tar ztf…” then put the file back?
Dennis Ndubi (13:33:03) (in thread): > My problem is solved.. thank you
Busayo Samuel (she/her) (13:33:36) (in thread): > No problem
Dennis Ndubi (14:35:00) (in thread): > What might be the problem, after running the R CMD, that is the output I am getting. i have removed the Rnw and committed the changes.. any directions@Madelyn Carlson@Andres Wokaty - File (PNG): image.png
{kind=link}
Emmanuel Taiwo devbird007 (21:21:32): > Hello guys, I have a problem, so this is what I am trying to do in a .Rmd file: > * Write several code chunks that create their own individual images > * Hide everything in these code chunks with include=FALSE > * Next, create a table with R Markdown syntax like this one:Authoring R Markdown Vignettes #Table > * Then, call these code chunks by their chunk-label into their specific row and column in the table. > I’ve been stuck at it for a while now, is it okay if I just use HTML for this type of occurrence in my file? > > > I’ve attached three images that represent: > 1. The section as displayed in the PDF file > 2. My attempt with HTML syntax > 3. The HTML code I used to generate my own attempt > Any help would be appreciated, I’ve been stopped at this stage for a day or two now trying to find the proper R Markdown syntax that could help. I’m going off now, but would continue with the issue when I wake up. > Thankssss@Andres Wokaty@Paul Villafuerte@Madelyn Carlson - File (PNG): Screenshot from 2023-03-19 02-17-35.png - File (PNG): Screenshot from 2023-03-19 01-55-37.png - File (PNG): Screenshot from 2023-03-19 02-08-04.png
{kind=link}
{kind=link}
{kind=link}
Andres Wokaty (23:56:00) (in thread): > The output on the left is showing that .Rnw is still there and it is using that to generate the PDF in the tar ball. Do you have 2 different directories for GOstats? On the left it says “c/Users/NDUBI” and on the right it is “c:/Users/NDUBI/OneDrive/Desktop/GOstats/vignettes”.
2023-03-19
Andres Wokaty (01:10:30) (in thread): > You might be able to uselayoutto help with this. Seehttps://statisticsglobe.com/r-layout-function-arrange-plots/. - Attachment (Statistics Globe): layout Function in R (3 Examples) | Arrange Grid of Plots in Base R > How to apply the layout function in R - 3 R programming examples - Complete R programming code in RStudio - Thorough information
Emmanuel Taiwo devbird007 (06:42:35) (in thread): > @Andres WokatyThanks, I went through the article and it was super helpful, I wish I had asked for help sooner really. Attached is my final work with thelayout()function. - File (PNG): Screenshot from 2023-03-19 11-41-18.png
{kind=link}
Sonali Kumari (10:29:54): > @Andres WokatyThere is a question mark in this pdfhttps://bioconductor.org/packages/devel/bioc/vignettes/GOstats/inst/doc/GOvis.pdfin first paragraph I have found out that there is no references related to this “” that’s why it is showing “?” here in pdf what should I do ? In this case .
Sonali Kumari (10:31:38) (in thread): > I tried using relevant reference for this but all the 7 references given in pdf are already used in the .Rnw file and there is no Reference left and also not relevant to this “GentGO2004a”.
Reece11 (12:46:36): > @Reece11 has joined the channel
Dennis Ndubi (14:13:26): > hello, is there a problem there, and what should I do to correct it… any directions will be appreciated. Thank you@Andres Wokaty - File (PNG): image.png
{kind=link}
Emmanuel Taiwo devbird007 (16:17:40) (in thread): > I don’t think there is a problem with it if that is really the size of the file
Emmanuel Taiwo devbird007 (20:48:26): > Hello everyone, the Summary section in my PDF file appears before the table-of-content, but all my attempts to replicate it in HTML always end up with it after the table-of-content, I could use some help with this, thanks.@Andres Wokaty@Paul Villafuerte - File (PNG): Screenshot from 2023-03-20 01-40-35.png - File (PNG): Screenshot from 2023-03-20 01-41-11.png
{kind=link}
{kind=link}
2023-03-20
Dennis Ndubi (03:36:33) (in thread): > okay
Dennis Ndubi (04:35:27): > hello,,, i need help to achieve this indentation(Or rather format it as one block), I have tried everything but I have not known the tricky, maybe I am missing some trick. Your assistance will be appreciated.@Andres Wokaty@Paul Villafuerte@Sonali Kumari@Emmanuel Taiwo devbird007the first frame is the original pdf, the second frame is my output. Thank you. - File (PNG): image.png - File (PNG): image.png
{kind=link}
{kind=link}
Sonali Kumari (05:48:16) (in thread): > Hello@Dennis NdubiI am not 100% sure but you can try to refer this for the problem you statedhttps://bookdown.org/yihui/rmarkdown-cookbook/indent-text.html.
Dennis Ndubi (06:13:14) (in thread): > i have tried it on the my Rmd file and it is not real working
Dennis Ndubi (07:23:55) (in thread): > i think i have managed to handle it
Sonali Kumari (07:54:08) (in thread): > ohk@Dennis Ndubi.. I am glad that your problem is solved.
Dennis Ndubi (07:57:26) (in thread): > Anytime@Sonali Kumari, and learn more in this field of open source and tech in general.. so incase you might have a project or an idea we can work on, holla @ me
Sonali Kumari (08:33:18) (in thread): > Sure.
Andres Wokaty (08:54:16) (in thread): > What is this the output of?
Aliyu Atiku Mustapha (09:05:55): > Good morning,@Andres WokatyPlease when you are free I have one last thing to ask before you review my work. > > There is a citation that doesn’t work on line 269 of first screenshot. Checking the Rnw file (second screenshot), it cites a GrossYellen, which i think is a book but the name is not right as it only mentions the surname of the authors (Gross & Yellen). Check third screenshot. What should I do? Thank you - File (PNG): image.png - File (PNG): image.png - File (PNG): image.png
{kind=link}
{kind=link}
{kind=link}
Dennis Ndubi (09:12:26) (in thread): > Running R CMD build for GOstats Package
Andres Wokaty (10:22:41) (in thread): > It might be better to get into the habit of copying pasting rather than screen shots as I can’t see the command you entered, the path, etc., so it’s really difficult to assess the problem. > > This is the build report for GOstats:https://bioconductor.org/checkResults/3.17/bioc-LATEST/GOstats/For windows,R CMD buildlooks likehttps://bioconductor.org/checkResults/3.17/bioc-LATEST/GOstats/palomino3-buildsrc.htmlDid you create GOstatsHyperG_cache?
Dennis Ndubi (10:35:54) (in thread): > Yes,, there is GOstatsHyperG_cache
Andres Wokaty (10:36:25) (in thread): > Why did you make GOstatsHyperG_cache?
Dennis Ndubi (10:38:36) (in thread): > It added itself to the vignettes folder … attached screenshot
Dennis Ndubi (10:38:41) (in thread): > should i delete it? - File (PNG): image.png
{kind=link}
Andres Wokaty (10:39:12) (in thread): > can you cut/paste the commands you are using?
Dennis Ndubi (11:00:07) (in thread): > After converting the GOstatsHyperG.Rnw file to Rmd file using the command > > pandoc -f latex -t markdown GOstatsHyperG.Rnw -o GOstatsHyperG.Rmd > > the files GOstatsHyperG.Rmd, GOstatsHyperG.html, GOstatsHyperG_case were automatically added,,, the I removed the Rnw file alone
Andres Wokaty (11:00:46) (in thread): > Good catch. I think there’s not an obvious solution to this. I see in the .bib file that there’s a GentGO2004B but no A version. It may be that we should ask the author what it should be, but I’m not sure when we can get a response. For right now, please continue working on the .Rmd and make a note of it in the PR so that it isn’t forgotten.
Andres Wokaty (11:08:35) (in thread): > R CMD buildcreates html but it creates the html in the/docpath. the html in your screen shot is in/vignettesso you must be generating them another way.
Andres Wokaty (11:08:40) (in thread): > that pandoc command doesn’t create html.
Andres Wokaty (11:09:43) (in thread): > I would remove the .html files from/vignettesand also/GOstatsHyperG_cache
Sonali Kumari (11:10:39) (in thread): > Thank you@Andres Wokaty. I will complete the rest of the task for .Rmd file and make a PR soon.
Andres Wokaty (11:11:26) (in thread): > I think this might be fine. In some cases, we can’t always make things appear as they do on the PDF.
Dennis Ndubi (11:12:15) (in thread): > okay thank you…lem do it
Andres Wokaty (11:31:14) (in thread): > Good catch.@Sonali Kumarialso has a similar problem. I decided to discuss with the core team about these citations. It might take a little time to get back. Please continue with the remainder of the vignette. As I mentioned to Sonali, Aliyu please document this issue in the PR so that it is not lost.
Aliyu Atiku Mustapha (11:34:26) (in thread): > Will do. So how long do you expect before hearing back from the team? Because I wanted to do another vignette as only one remain in the dashboard
Andres Wokaty (11:37:11) (in thread): > Generally, it takes some time as everyone has different opinions and we try to agree as a team what is the best course of action. > > We still need to finish the rest of your review before you get another vignette
Aliyu Atiku Mustapha (11:38:09) (in thread): > Alright. > I think I have covered everything requested of me. Please I have a look when you are free. > Thank you
Dennis Ndubi (11:42:28) (in thread): > it has worked fine… thank you@Andres Wokaty
Andres Wokaty (12:27:02) (in thread): > Can you add the following in the right format to the .bib file: > > Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000 May;25(1):25-9. doi: 10.1038/75556. PMID: 10802651; PMCID: PMC3037419. > > You’ll need to update the corresponding reference in the .Rmd. > > Here’s a general reference on bibliographies if you need it:https://bookdown.org/yihui/rmarkdown-cookbook/bibliography.html. - Attachment (bookdown.org): 4.5 Bibliographies and citations | R Markdown Cookbook > This book showcases short, practical examples of lesser-known tips and tricks to helps users get the most out of these tools. After reading this book, you will understand how R Markdown documents are transformed from plain text and how you may customize nearly every step of this processing. For example, you will learn how to dynamically create content from R code, reference code in other documents or chunks, control the formatting with customer templates, fine-tune how your code is processed, and incorporate multiple languages into your analysis.
Sonali Kumari (13:56:14) (in thread): > ohk . Thank you@Andres Wokaty
Sonali Kumari (15:19:23) (in thread): > @Andres WokatyI have noticed in .Rnw file the “GentGO2004a” is used instead of GentGO2004B, I think this could be also a reason of that “?” in pdf . Please see this.
Andres Wokaty (15:23:15) (in thread): > I couldn’t find that paper on a cursory search, so I asked someone who worked on graph and they gave me the reference I gave to you.
Emmanuel Taiwo devbird007 (15:25:03) (in thread): > Okay, thanks
Sonali Kumari (15:27:30) (in thread): > Also@Andres WokatyI found out similar information given by you in the .bib file do i still need to add another ? - File (PNG): 2023-03-21.png
{kind=link}
Andres Wokaty (15:29:06) (in thread): > No, don’t add another. link to that one.
Sonali Kumari (15:29:19) (in thread): > ohk. Thanks for clarification:blush:
Dennis Ndubi (17:42:52): > Hello, @ line 298 there is a package (Yeast) i am wondering whether it is a Bioconductor package because when I refer to it using the Biocpkg, it is not available on the landing page. What should I do@Andres Wokaty@Paul Villafuerte. But there is yeast.dbo which is annotation package still. - File (PNG): image.png - File (PNG): image.png - File (PNG): image.png
{kind=link}
{kind=link}
{kind=link}
2023-03-21
MaryJones (06:03:15): > @Andres WokatyGood day everyone:
Oluwabukola Bamigbade (06:07:00): > @Oluwabukola Bamigbade has joined the channel
MaryJones (06:33:06): > @Andres WokatyThank you very much for you great advice, I did as instructed and using all the points you gave, followed them all. So I had to convert to a .tex file and then from .tex-file, pandoc created an .Rmd using the .tex file. so I am cleaning up the .Rmd file and inserting the missing paragraghs , the table was did not display correctly so I had to create a new one. please take a look at the resulting .Rmd I have now and kindly peruse it. Thank you all, I appreciate:pray: - File (MPEG 4 Video): My new .Rmd-file and its output.mp4
MaryJones (06:35:39): > @Emmanuel Taiwo devbird007Hello, yes thank you, I am making some progress.:pray:, The mentors already helped me out. thanks a lot.
Paul Villafuerte (08:33:19) (in thread): > I am aware of this issue. Please insert as “YEAST” in bold for now and continue with the rest of the conversion.
Dennis Ndubi (08:34:03) (in thread): > Okay..
Madelyn Carlson (10:00:01): > Hi all - as a reminder, we are hosting the Sweave2Rmd-Outreachy office hour tomorrow at 11-11:45AM ET (Zoom link). We answer questions about the project and contribution tasks. If you’re unable to make it, we have another office hour session next week on Wednesday, March 22, 10-10:45AM ET. Hope to see you there!
Sonali Kumari (13:26:18): > @Andres Wokaty@Paul VillafuerteI want to ask if we have to answer any “Community-specific Questions” for Outreachy final application? - File (PNG): 2023-03-21 (2).png
{kind=link}
Andres Wokaty (13:59:34): > @Sonali KumariThanks for asking. For your final application, could you please answer the following: > > * Why are you applying to this project? > * What do you hope to get out of your participation?
Sonali Kumari (14:10:56) (in thread): > Ohk Thank you@Andres Wokaty.
2023-03-22
MaryJones (02:40:12): > @Andres WokatyGood day everyone, please here is the final output document for your perusal. Please may you kindly check it and advise on anything else you may want me to remove or add. And please advice if I may go ahead and make a PR as well as publish. thank you all,:pray: - File (Binary): Using Chromosome Bands as Categories.webarchive - File (MPEG 4 Video): Kemickyz(MaryJones)_Final .Rmd Output.mp4
Dennis Ndubi (04:10:45) (in thread): > Hello,@Paul Villafuerteat your free time, Would you kindly let me know if there are any changes I should make to the pr, since I have done the previous correction asked
Emmanuel Taiwo devbird007 (04:56:46): > Hi@MaryJones, just off the bat from what I can see, if you have several lines that are like this in the PDF file: > > text1 > > text2 > > text3 > > They should actually be represented like this in your R Markdown file:
text1 text2 text3
This is because they are code chunks. They also usually come with extra parameters in the code chunk header that it appears you have removed from your own document. I’d advise you go through theAuthoring R Markdown vignettespage to familiarize yourself with some of these things.
I also think you should use the
pandoc -f latex -t markdown file.Rnw -o file.Rmdcommand to convert from .Rnw to .Rmd, this will preserve the code chunk parameters and then you can focus on manually editing the chunks.Hopefully this also helps anyone who may be having a similar problem.
Andres Wokaty (09:08:18): > @MaryJonesYou should make your PR. I know this wasn’t an easy vignette. It’s easier to provide feedback on PRs on github rather than on slack.
Sonali Kumari (09:26:17): > @Andres Wokaty@Paul Villafuerte@Madelyn Carlsonthis is my LinkedIn post regarding my very first contribution in open source, I would love to connect with you all in LinkedInhttps://www.linkedin.com/posts/sonali-kumari-89917622a_replacinggenomicfilesrnwtogeno[…]624027303936-jKSB?utm_source=share&utm_medium=member_desktop - Attachment (linkedin.com): Sonali kumari on LinkedIn: Replacing_GenomicFiles.Rnw_to_GenomicFiles_Rmd by sonali8434 · Pull… > Hey connections!!! > > The past two weeks have been really exciting and swotting for me since I have started my very first journey of my Open Source contribution…
Madelyn Carlson (10:00:01): > Friendly reminder that our Sweave2Rmd-Outreachy office hour starts soon. Feel free to join for the 11-11:45AM ET session,Zoom link here
khadijah (10:35:43) (in thread): > Welldone:sparkles::sparkles::sparkles:
Paul Villafuerte (15:29:06) (in thread): > I will. Do this evening
2023-03-23
Dennis Ndubi (01:02:22) (in thread): > hello, i have removed the extra CR, and i would like to know which IDE i would use to note the presence of the carriage returns and remove them as early as possible to ease your work, thank you.
Andres Wokaty (10:51:50) (in thread): > RStudio has setting to show white space characters (Tools > Global Options. In Global Options, Code > Display you’ll see a checkbox to show white space characters)
Aliyu Atiku Mustapha (11:16:48) (in thread): > :eyes:Aren’t we behind on the time?
Madelyn Carlson (11:19:57) (in thread): > Hi@Aliyu Atiku Mustaphawe switched office hours from Thursday to Wednesday this week because folks during our session last week helpfully pointed out that that our office hours overlapped with BugSigDB office hours,see my Slack message here. Here are our notes from the session yesterday if you want to check out what we talked about:https://community-bioc.slack.com/archives/C037LN1HQDB/p1679500044061979?thread_ts=1679493603.703949&cid=C037LN1HQDB - Attachment: Attachment > Thank you to folks on our office hours today pointing out that our office hours overlap with BugSigDB office hours. In case y’all are interested in attending both sessions, we’re switching the Sweave2Rmd office hour in 2 weeks from Thursday, March 30 to Wednesday, March 29 at 10-10:45AM ET. So here is the schedule for the next two weeks. > • Wednesday, March 22, 11-11:45AM ET (Zoom link) > • Wednesday, March 29, 10-10:45AM ET (Zoom link) > Hope to see you there. - Attachment: Attachment > Key points & notes from our office hours session today: > • If you’ve made one or multiple PRs, some merged and some unmerged, you may link to both in the final Outreachy application, if you choose to complete an application for the Sweave2Rmd project. Or, if you’ve made one PR that is unmerged, you should post a link to that in the application. > • In the final Outreachy application for the Sweave2Rmd project, you will be asked to complete community questions. Those two questions are (1) why are you applying to this project? and (2) What do you hope to get out of your participation?
Madelyn Carlson (11:20:53) (in thread): > We’re meeting again next Wednesday, 10-10:45AM if you’re available to join! In the meantime, feel free to post any questions you have in Slack
Aliyu Atiku Mustapha (11:22:00) (in thread): > Oh, I thought the message was posted today. Just noticed. See you next week then. Thank you
Madelyn Carlson (11:23:52) (in thread): > All good! See you next week
Sonali Kumari (12:01:16): > @Andres WokatyWhile Running R CMD build GOstats I am not getting any error it builds perfectly but while running this command tar ztf GOstats_2.65.0.tar.gz | grep doc/GOvis it does not show anything . Do you know the reason behind it ?These 2 pictures are from my project terminal. - File (PNG): 2023-03-23 (2).png - File (PNG): 2023-03-23 (1).png
{kind=link}
{kind=link}
MaryJones (19:02:54) (in thread): > @Andres WokatyGood everyone,
MaryJones (19:05:39) (in thread): > @Andres Wokatythank you very much, so I clone the original repo from Bioconductor’s site directly because there was no GitHub url in the documentation for me to fork
MaryJones (19:08:17) (in thread): > @Andres WokatySo I created a GitHub repo for review for now, here is the link :https://github.com/kemicky/file-Rmd
Andres Wokaty (19:09:20) (in thread): > I see that. I think it’s better to fork Category, clone it to your computer, then copy the right files from file-Rmd to the cloned Category repo.
Andres Wokaty (19:10:31) (in thread): > The Fork button is on the top right (there are 4 buttons there) - File (PNG): Screenshot from 2023-03-23 19-08-17.png
{kind=link}
MaryJones (19:12:06) (in thread): > @Andres WokatyI created for the time being pending your further instructions if I must push it to biconductor site and please may you advice me on how to make the pull request on Bioconductor site after pushing the files.And may I start to convert the second one Category.Rnw file while I await your further instructions. Thank you all,:pray:
Andres Wokaty (19:13:55) (in thread): > Unfortunately, you can’t do a pull request with that repository.
Andres Wokaty (19:15:05) (in thread): > I can give you some guidance to fork/clone, copy the right files, then do the pull request
MaryJones (19:15:26) (in thread): > Ok may I get this GitHub url , then I fork the repo, not a problem, I will do it now by morning my time I guess the PR will be ready.
MaryJones (19:17:04) (in thread): > Ok I find it and I will be done by morning my time for your review. Thank you very much,:pray:
Andres Wokaty (19:17:35) (in thread): > https://github.com/Bioconductor/Categoryis that what you were looking for?
Andres Wokaty (19:19:04) (in thread): > Please only make the changes to the necessary files as the hope here is that we can merge your changes into Bioconductor.
MaryJones (19:19:46) (in thread): > Yes please, thank you very much,:pray:
Andres Wokaty (19:20:37) (in thread): > good luck:slightly_smiling_face:
MaryJones (19:21:07) (in thread): > Ok, I will do so, any file you want me to remove after the review I will remove asap, thank you very much,:pray:
MaryJones (19:32:52) (in thread): > @Andres WokatyI have forked the repository as adviced, may I clone directly from the one I forked to my GitHub?, please advice ,, Thank you - File (PNG): Category repo i forked.png
{kind=link}
Andres Wokaty (19:35:01) (in thread): > You clone the forked repository on your computer.
MaryJones (19:35:20) (in thread): > OK, they are just as it is in the pdf, the ones in the r markdown are missing. Thank you. I will fix them after the reviews, thanks a lot,:pray:
MaryJones (19:36:23) (in thread): > Ok, I will do that now , I am very grateful for your wise guidance,:pray:
Andres Wokaty (19:58:04) (in thread): > The last time I saw this behavior, it was because the set up in the DESCRIPTION file wasn’t correct, but the DESCRIPTION looks okay. I confirmed that the package in our git repository builds the PDFs. I would confirm that it is still building the PDFs (just dogrep doc). If it isn’t, maybe try removing your .Rmd file out and see if it will build the PDFs that way then we might know if it is something in the .Rmd file. I’ll try to troubleshoot tomorrow as it is getting late here.
MaryJones (23:22:37): > @Andres WokatyGood day everyone, I am done with the PR, please may you kindly find the Github link here:My file-Rmd Github linkand the PR link :My Pull Request linkfor your perusal and review, thank you all,:pray: - Attachment: #4 File rmd
2023-03-24
Sonali Kumari (02:58:07) (in thread): > ohk I will do all the steps advised by you and share the results with you.
Dennis Ndubi (03:50:12) (in thread): > Thank you@Andres Wokaty
Dennis Ndubi (03:53:20) (in thread): > And i would like to know whether i have done everything well since i have made the changes requested previously@Andres Wokaty@Paul Villafuerte..i am eager to see my pr merged:sunglasses:
Sonali Kumari (05:05:30) (in thread): > @Andres Wokatyyes I have done all the steps asked by you first of all I have done grep doc only without removing .Rmd file and this is the output - File (PNG): 2023-03-24 (3).png
{kind=link}
Sonali Kumari (05:10:47) (in thread): > And this is the output after removing .Rmd file and adding .Rnw file as said by you I am getting that there might be problem in .Rmd file but I have checked it and unable to find out whare exactly the problem is I guess you might check my check my .Rmd file and might find out why there is a problem. - File (PNG): 2023-03-24 (4).png
{kind=link}
Sonali Kumari (06:06:38) (in thread): > Hello@MaryJonesI am also a learner and I can understand your situation well, I have also done lot of mistakes in my first PR good luck for your PR one small piece of advice from my side we should name our PR in a correct way so that the maintainer could understand the reason why we are making a PR. So according to your PR you can rename your PR as Conversion_of_ChromBand.Rnw_to_ChromBand.Rmd
Sonali Kumari (06:08:43) (in thread): > I advise you to please rename your PR you can also rename your branch now it is file-Rmd it should be ChromBand-Rmd. I hope it helps have a good day.
Sonali Kumari (08:45:59) (in thread): > @Andres WokatyI found out the problem and solved it actually the one line that is missing in my YAML file that is why R CMD Check is not showing the correct result. I have made the changes and now all my checklists are done . Here is the output of R CMD build and check after correcting the error . Thank you for always guide us in right path. - File (PNG): 2023-03-24 (5).png
{kind=link}
Andres Wokaty (10:00:43): > Hi, I just want to remind everyone to keep links to your PRs inhttps://github.com/Bioconductor/sweave2rmd/issues/63. While you wait for your review, please use the checklist inhttps://bioconductor.github.io/sweave2rmd/articles/contribute.html#review-a-pull-requestto review your work and make adjustments. Please be patient waiting for your review as we are working with everyone to make sure your PR get merged. - Attachment (bioconductor.github.io): Contribute > Learn how to contribute to our project.
Andres Wokaty (10:04:31) (in thread): > What line was missing in the file? And would you like to add a section inhttps://github.com/Bioconductor/sweave2rmd/blob/main/vignettes/troubleshooting-faq.Rmdabout it? You can include this as a contribution if you decide to do it.
Sonali Kumari (10:23:47) (in thread): > % this line is missing if we miss one this signal line then we face a problem actually which i was getting earlier. so can I add this ? in the document FAQ section? I think this is a very stupid mistake done by me can I still make a contribution for this error?
Andres Wokaty (10:25:56) (in thread): > But I think it’s an easy error anyone can make that causes a huge problem that is difficult to troubleshoot. It would be great if you could add this to the documentation. Please include a description of the problem, how to troubleshoot, and your solution as one possible solution in the Q and A style format that is used.
Sonali Kumari (10:26:00) (in thread): > I can explain about the importance of YAML header and the work each line does
Andres Wokaty (10:26:17) (in thread): > sounds good
Sonali Kumari (10:26:49) (in thread): > Ohk Thank you Jennifer I would love to do that.
Oluwabukola Bamigbade (10:53:16): > Hi@Andres Wokaty@Madelyn CarlsonI have been encountering the same error code trying to markdown. The error message “unexpected end of input” suggests that there may be an issue with the contents of the Workflow.Rnw file. Specifically, it seems that there may be a problem with the file’s formatting or syntax at or before line 520. > I have double-checking the contents of the Workflow.Rnw file, particularly in the area surrounding line 520, to ensure that the file is correctly formatted and free of any syntax errors or other issues but I can find any but I keep running into the same error message all over again. Please advice - File (JPEG): e3e712bd-b303-47e4-8fee-44dd9c3c3133
{kind=link}
Andres Wokaty (10:56:48) (in thread): > Can you share the link to the package you’re working on? This happens when pandoc is having difficult parsing a particular section. Usually, I have to remove chunks of code from the bottom of the .Rnw file then run pandoc to find the problem area. I repeat this until I find the section. Then I add code back in and leave out the problem part so I can make pandoc do most of the conversion work. After creating the .Rmd file, I add back in the problem section in R Markdown format.
Andres Wokaty (10:57:35) (in thread): > I think the last time I experienced this, the figures in the .Rnw were problematic for pandoc
Oluwabukola Bamigbade (10:57:45) (in thread): > https://bioconductor.org/packages/devel/bioc/html/chipseq.html - Attachment (Bioconductor): chipseq (development version) > Tools for helping process short read data for chipseq experiments
Oluwabukola Bamigbade (10:58:55) (in thread): > @Andres WokatyI have shared the link to the packageI’mworking on
Andres Wokaty (11:15:45) (in thread): > I think Pandoc is having an issue with > > -<<>>= > -peakSummary$inPromoter <- peakSummary %over% promoters > -xtabs(~ inPromoter + change, peakSummary) > -@ > > Remove this chunk of code and try converting with pandoc then add it back in to the .Rmd
Oluwabukola Bamigbade (11:16:50) (in thread): > Okay.I’lldo that now. Thank you
Oluwabukola Bamigbade (11:20:14) (in thread): > It worked. Thank you:relaxed:
Sonali Kumari (14:58:11) (in thread): > @Andres Wokatyhere is my PR Link-https://github.com/Bioconductor/sweave2rmd/pull/67for the Q and A should I link it herehttps://github.com/Bioconductor/sweave2rmd/issues/63also or not? - Attachment: #67 Updated troubleshooting-faq.Rmd - Attachment: #63 Replace a Sweave Vignette With an R Markdown Vignette
Andres Wokaty (15:01:29) (in thread): > Let’s not include it since it’s not a vignette. As I mentioned, you can still list this as a contribution for this project since it came up during your other contribution task. I’ll likely review it next week since it’s almost end of day here. Thank you for working on it.
MaryJones (15:03:19) (in thread): > @Sonali KumariHello, Ok thank you so much, Ok then I will do that. Thank you so much for your helpful advice.
Sonali Kumari (15:03:23) (in thread): > ohk, thank you so much.
MaryJones (22:52:58): > @Andres WokatyHello, I have removed the other files,I dot understand what you mean by generating the image files and table. I converted the files again and the images are generated just the same as embedded pdf ..please can you explain what to do ,I have exhausted every resolution I could find, even if I copy back those code the rmd doesn’t compile. the only solution I keep arriving at is it creates the images like it is and I knit it in the r code. please kindly advice how you want me to solve this issue. thank you all,:pray:
2023-03-26
Dennis Ndubi (08:41:03): > hello, can someone work on another pr while another is in review@Andres Wokaty@Paul Villafuerte
Emmanuel Taiwo devbird007 (09:07:24) (in thread): > Hi Dennis, You can refer to Jen’s commenthereon the issue. Your PR first needs to be merged before you can work on another vignette. - Attachment: Attachment > Did you finish the genefilter vignette? If you finished it, please put a link to your PR in https://github.com/Bioconductor/sweave2rmd/issues/63 so we can update the board. Everyone has to finish the review on their first PR before getting another assignment. Because no one has done a perfect PR, I am recommending that everyone review their PR using the checklist at the bottom of the contribution document. This may reduce the amount of work you need to do during the review.
Dennis Ndubi (09:17:16) (in thread): > i understand
Dennis Ndubi (09:50:35): > when making the final application, there is a field that requires outreachy internship project timeline and it says to work with your mentor to provide a timeline of the work you plan to accomplish on the project and what tasks you will finish at each step. Could you@Andres Wokaty@Madelyn Carlsonmake some clarification here how one should go about it? - File (PNG): image.png
{kind=link}
Paul Villafuerte (13:25:39) (in thread): > @Dennis NdubiThere is one more change to your PR. Please review. Thanks!
Dennis Ndubi (13:32:04) (in thread): > Hello@Paul Villafuerte, i have made the change by adding the needed CR and commited it. Kindly check. Thanks
Paul Villafuerte (13:33:09) (in thread): > @Dennis NdubiYour PR is approved. Thanks!
Dennis Ndubi (13:38:29) (in thread): > @Paul VillafuerteAnytime, Now I guess I have the green light to select another PR from the board and continue contributing as I polish my skill…or should i wait until the status changed to accepted at the project board
Paul Villafuerte (13:39:16) (in thread): > Please go ahead and choose another PR.
Dennis Ndubi (13:43:27) (in thread): > @Paul Villafuertethis is the remaining unasigned PR.. OrganismDbi/vignettes/OrganismDbi.Rnw(https://github.com/Bioconductor/OrganismDbi).. I can take it
Dennis Ndubi (13:47:46) (in thread): > @Paul Villafuerteand can you kindly clarify this for me(in the final application)https://community-bioc.slack.com/files/U04SWP221LP/F050PAPEENM/image.png - File (PNG): image.png
{kind=link}
Dennis Ndubi (14:39:32) (in thread): > @Paul Villafuertehello, this is about the YAML file.@Sonali Kumariadvised me that it is good to add links for intext citations and i think i should make them, should i go ahead?
Dennis Ndubi (15:04:46): > Hello@Andres Wokaty@Paul VillafuerteI am just asking in general, in the YAML metafile specifically the date line, which is more appropriate, a static date or a dynamic system-generated date
khadijah (22:38:46) (in thread): > Hi you can use"format(Sys.time(), ‘%B %d , %Y’)"
khadijah (22:39:00) (in thread): > It has already been updated in the docs
2023-03-27
Paul Villafuerte (01:29:02) (in thread): > I don’t think they are part of the original pdf. We want to keep the Rmd’s as close to the original as possible.
Dennis Ndubi (01:32:19) (in thread): > okay, thank you for clarification
Emmanuel Taiwo devbird007 (02:42:35) (in thread): > Hello@Paul Villafuerte, I noticed you were assigned to my PR on theProject Board, Here is thelinkto the PR, maybe you could go over it whenever you are less busy. I have also taken the time to go over it myself with the checklist. - Attachment: #3 Conversion of RBGL.Rnw to Rmd
Dennis Ndubi (02:47:38) (in thread): > thank you
khadijah (04:57:23) (in thread): > You are welcome
Sonali Kumari (13:37:58) (in thread): > @Andres Wokaty@Paul Villafuerte@Madelyn CarlsonI am also struggling with the same question that requires collaboration with a mentor. Could you please provide some guidance on how to approach the question effectively?
Andres Wokaty (15:08:11) (in thread): > For the timeline, please look at the project description onOutreachy.organd try to identify important tasks and group them into milestones. This is also something that will be discussed and worked on together during the internship, so we’re looking to see how you organize. It would be great if you could mention any commitments during the internship round.
Sonali Kumari (20:05:31) (in thread): > Ohk@Andres WokatyThank you so much
2023-03-28
Dennis Ndubi (04:40:14) (in thread): > Thank you@Andres Wokaty
MaryJones (10:48:08): > @Andres WokatyGood day everyone, I did the corrections and here is the table generated, please advice if its ok. thank you, or must I style it like this ? please advice. thank you all - File (PNG): Screen Shot 2023-03-28 at 4.42.38 PM.png - File (PNG): Screen Shot 2023-03-28 at 4.46.58 PM.png
{kind=link}
{kind=link}
Andres Wokaty (10:53:53) (in thread): > This is output, correct? So you don’t style output.
MaryJones (10:57:16) (in thread): > Yes please, the output is correct, but I dont know how to remove the ##, that is the generated output.
Andres Wokaty (10:58:30) (in thread): > You can leave it if it is generated
MaryJones (10:58:35) (in thread): > And with the images must I add the ignored codes that are commented out ?
Andres Wokaty (10:59:28) (in thread): > I don’t understand. Can you be specific by pointing me to an example/lines of code?
MaryJones (11:09:36) (in thread): > Here is the screenshot, I notice it is only line 123 and 124 are the ones needed for generating the image. can I remove the other commented out ones in green ?, please peruse and advice, thank you:pray: - File (PNG): Screen Shot 2023-03-28 at 5.06.01 PM.png
{kind=link}
Andres Wokaty (11:10:39) (in thread): > I believe this is as it is in the .Rnw, so let’s keep it. It still goes inside the code block.
MaryJones (11:13:06) (in thread): > yes ,I now put them the code blocks as you advised and the images and tables are generated automatically now. thank you very much for your wise guidance, I will do them as instructed,:pray:
MaryJones (12:11:20) (in thread): > @Andres WokatyI am done, must I remove all the Rnw files including the category.Rnw ?
Madelyn Carlson (12:19:33): > As a reminder, we are hosting the Sweave2Rmd-Outreachy office hour tomorrow at 10-10:45AM ET (Zoom link). We answer questions about the project and contribution tasks. Hope to see you there!
MaryJones (12:29:47) (in thread): > Here is a screenshot of the files left, please advice if there is anyone you want to me to remove before PR. The case folder is created automatically I guess because some of the codes have their cache property turned on, please advice if I must delete it. Thank you
MaryJones (12:30:19) (in thread): - File (PNG): Screen Shot 2023-03-28 at 6.21.32 PM.png
{kind=link}
Andres Wokaty (12:40:15) (in thread): > Remove Chromband_cache
Andres Wokaty (12:41:48) (in thread): > You remove any files you added except for ChromBand.Rmd. You also remove ChromBand.Rnw.
Andres Wokaty (12:42:50) (in thread): > Category.Rnw is a separate vignette so leave that.
MaryJones (12:54:36) (in thread): > ok thank you, very much. I am on it immediately,:pray:
MaryJones (13:42:29): > @Andres WokatyHello everyone
MaryJones (13:44:22): > @Andres WokatyI pushed a new correct branch chromBand-Rmd , so can I delete the old branch that is wrongfully named file-Rmd ? before making the PR, please advice ,:pray:
Andres Wokaty (13:53:08) (in thread): > You can close the old one and open a PR for the new one, which ishttps://github.com/Bioconductor/Category/compare/devel…kemicky:Category:chromBand-Rmd. I just want to be careful about doing this because we lose the history of when we first started working on the PR together. So we will stick to this new PR. > > Let’s also keep all discussion there. I receive notifications about PRs I am associated with. > > Lastly, I still see extra files in the new branch.
MaryJones (13:58:34) (in thread): > Ok, yes please. Thank you, I will do as you instructed. I appreciate
MaryJones (14:24:22) (in thread): > Done, made the PR. please may you kindly review. thank you very much for your patience and wise guidance, I am very grateful:pray:
MaryJones (15:14:02): > @Andres Wokatythank you for your review and.. I have done the check and the tar.gz if generated automatically see here. So Must I add it to the folder ? - File (Gzip): Category_2.65.0.tar.gz
Andres Wokaty (15:15:54) (in thread): > No, don’t add any other files. The reviewer will doR CMD buildon the package to verify that the vignette builds.
MaryJones (15:16:41) (in thread): > ok. thank you:pray:
MaryJones (15:17:41) (in thread): > @Andres WokatyPlease while I wait for the response may I be assigned a new one, all the ones on the project board are assigned , thank you, :pray:
Andres Wokaty (15:18:23) (in thread): > I’ve asked everyone to wait until their first PR is merged.
MaryJones (16:47:01) (in thread): > Ok, thank you so much.:pray:
2023-03-29
Emmanuel Taiwo devbird007 (00:55:02) (in thread): > Hi@Paul Villafuerte, I have carefully implemented your instructions, and you can now continue the review process.
Madelyn Carlson (09:00:04): > Friendly reminder that our Sweave2Rmd-Outreachy office hour starts soon. Feel free to join for the 10-10:45AM ET session,Zoom link here
Sonali Kumari (09:05:36): > Hello@Andres WokatyI have tried many citation style out of which I found one most similar which is biological-psychology so, we can use either biological-psychology.csl or bioinformatics.csl you can compare both of them from the screenshot attached and also the original pdf-https://bioconductor.org/packages/release/bioc/vignettes/GOstats/inst/doc/GOvis.pdf
Sonali Kumari (09:06:17) (in thread): > this is the output of biological-psychology.csl - File (PNG): 2023-03-29 (5).png
{kind=link}
Sonali Kumari (09:07:15) (in thread): > And this is the output of bioinformatics.csl which one should I use in .Rmd file? - File (PNG): 2023-03-29 (6).png
{kind=link}
MaryJones (09:44:04): > @Andres WokatyHello everyone , I ran the R CMD BUILD command for the review, there is no error and here is the result for your perusal: I am still waiting for the reviewers response, thank you all,:pray: - File (PNG): Screen Shot 2023-03-28 at 11.33.52 PM.png - File (PNG): Screen Shot 2023-03-28 at 11.34.30 PM.png - File (PNG): Screen Shot 2023-03-28 at 11.35.58 PM.png - File (PNG): Screen Shot 2023-03-28 at 11.36.35 PM.png
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Emmanuel Taiwo devbird007 (09:53:04) (in thread): > That’s super nice.
Andres Wokaty (10:01:47) (in thread): > Please be patient regarding reviews. The most important thing is it is on the board so eventually we will get to it. > > BTW, that looks likeR CMD check, which is also important. You want to make sure you can doR CMD buildthen grep for the compiled html in the tar ball.
MaryJones (10:18:23) (in thread): > Yes please, I did that too and**** it created the a folder doc and with several files the original .Rmd, an html format and an .r format and a tarGZip automatically. I observed the R CMD check, does both: 1. checks the .Rmd file for errors and 2. builds the .RMD, if there is no error.**** - File (Gzip): Category_2.65.0.tar.gz - File (HTML): ChromBand.html - File (R): ChromBand.R
Andres Wokaty (10:20:51) (in thread): > It’s good that you tested and it works. The reviewer will do the same tests.
MaryJones (10:24:29) (in thread): > ok
Andres Wokaty (10:25:38) (in thread): > Did you remove extra files?
MaryJones (10:27:32) (in thread): > Yes I did as you instructed, except the Category.Rnw you advised I must not remove, please check.
Andres Wokaty (10:28:43) (in thread): > So I just took a peek at your PR and there are extra files that should be removed. They are hidden files:https://github.com/Bioconductor/Category/pull/5/files
MaryJones (10:32:16) (in thread): > which ones, Please, Identify?, those came with the files, they are git files
Andres Wokaty (10:33:05) (in thread): > Your commits should touch the DESCRIPTION, .Rmd, and .Rnw files. Anything else is extra and should not be included in the PR.
MaryJones (10:38:49) (in thread): > I did do all that, check the commit history and I didn’t add any files. Nothing extra. I dont know what extra files you talking about , I no control over those git files, those are added when one is using Git. So please point out to me the extra files I should remove, thank you
Andres Wokaty (10:41:02) (in thread): > biological-psychology.csl does look closer but not perfect and it wouldn’t be the right publication. Let’s bioinformatics.csl, since I know one of the GOstats vignettes was published in that journal.
Andres Wokaty (10:41:27) (in thread): > When you look athttps://github.com/Bioconductor/Category/pull/5/files, it shows you all the files that are touched in the PR
Andres Wokaty (10:42:25) (in thread): > According to that, the following files are included: .DS_Store, .Rbuildignore, .gitignore, Category.Rproj, DESCRIPTION, ChromBand.Rmd, ChromBand.Rnw.
Andres Wokaty (10:43:19) (in thread): > .Rbuildignore and Category.Rproj come from using RStudio I believe, .gitignore is a git file, and I believe .DS_Store comes from macs?
Sonali Kumari (10:43:29) (in thread): > Ohk@Andres WokatyThank you so much:blush:..
Andres Wokaty (10:44:23) (in thread): > For Bioconductor packages, we don’t keep these files in the repository because they can be specific to the user or they’re unnecessary for the package itself.
Andres Wokaty (10:44:37) (in thread): > Please remove .DS_Store, .Rbuildignore, .gitignore, Category.Rproj.
Andres Wokaty (10:45:39) (in thread): > Just to clarify a little more by ‘touched’, I mean they are edited in some way, which can mean modified, removed, or added.
MaryJones (10:47:17) (in thread): > Those are not extra files , they are needed settings for Git to push files and that is why they are hidden, if I remove them all the entire files are corrupt. Anyone who use git or GitHub know this
Andres Wokaty (10:48:28) (in thread): > They are extra files. Removing them from your PR will have no impact on other files.
Andres Wokaty (10:49:27) (in thread): > You can still keep them on your computer, but they don’t belong in the PR.
MaryJones (10:49:38) (in thread): > as you can see those file are written with a prefix of . , meaning those are setting important files used by git to track those files , I didn’t write them.
Andres Wokaty (10:50:14) (in thread): > Yes, I know you didn’t write them. Some of them get automatically generated.
MaryJones (10:50:20) (in thread): > They do, why are you trying to stress me beyond limit ?, please
Andres Wokaty (10:50:59) (in thread): > I am not trying to stress you and I am sorry if you are stressed, but I am telling you how to improve your PR so that it can be merged.
MaryJones (10:52:17) (in thread): > You and I know those file are settings and there is no way I can do PR or version control without those files been added. everybody know this, removing it from my PR will make my branch not merge, you know this.
MaryJones (10:52:49) (in thread): > But the files is even confirmed by GitHub that they can merge.
Andres Wokaty (10:54:04) (in thread): > Ok, let’s take a step back and cool down.
Andres Wokaty (10:56:55) (in thread): > Here’s another PR I did awhile back to convert a vignette in the package zlibbioc:https://github.com/Bioconductor/zlibbioc/pull/2/files. You can see exactly the files that are touched.
MaryJones (10:57:17) (in thread): > Thank you very much.
Andres Wokaty (10:57:56) (in thread): > I’m not going to guide you in the wrong direction. My goal is the same as yours: to get your PR merged.
Andres Wokaty (11:00:37) (in thread): > If you look at the zlibbioc PR, you see it is merged and only the DESCRIPTION, Usingzlibbioc.Rnw, and Usingzlibbioc.Rmd files are touched.
Aliyu Atiku Mustapha (11:02:39): > Hi,@Madelyn Carlson@Andres Wokaty@Paul Villafuerteso here is the screenshot of my question related to flowcharts > First screenshot is the converted vignette showing the code and the output on the right side(error) > Second screenshot is the pdf file showing what the flowchart should look like > Third is what code i managed to create using DiagrammeR package and the viewer to the right showed what my code generated in the HTML file. > Obviously, more work to be done to make it more similar to what the pdf is showing like the Keys having a different font color and compartmentalized from the main object, but reading the DiagrammeR package documentation on their website, I don’t think it has all those capabilities. > I am taking a look at the dagitty suggested, maybe I can. > Lastly, Is there a reason where adding ‘fig.small = TRUE’ to my code chunk isn’t working to make the figure small? - File (PNG): image.png - File (PNG): image.png - File (PNG): image.png
{kind=link}
{kind=link}
{kind=link}
MaryJones (11:14:40) (in thread): > I dont understand anymore, I will just wait and see, if merges or not and whatever the review says, I have done everything instructed and I given it my all, I have so tireless on these file day and night for weeks. I guess I will try and rest bit. I am sure you can see the history of the files, I worked on there and Thank you for all your patience, I am very grateful,:pray:
Sonali Kumari (11:18:25) (in thread): > Also@Andres Wokatyjust asking for confirmation we have to add bioinformatics.csl in our branch (PR). In order to use this since in my computer it is necessary to add bioinformatics.csl in the vignette. ? Please confirm..
Andres Wokaty (11:18:52) (in thread): > yes, add this file to the PR
Sonali Kumari (11:19:09) (in thread): > Ohk thank you so much ..
Emmanuel Taiwo devbird007 (11:19:15) (in thread): > For your last question, I believe sometimes the effect offig.smallcan be only minimal on the Figure. You can try using the parametersfig.widthandfig.heightwhich would be equated to a number of your choice to try and get the desired output.
Aliyu Atiku Mustapha (11:21:45) (in thread): > Yes, fig.small didn’t seem to have any effect whatsoever, but because BiocStyle has said defined parameters, I didn’t think setting a specific defined figure height/width was a good idea. > Thanks
Emmanuel Taiwo devbird007 (11:24:49) (in thread): > You could also look through the Figures section of theAuthoring R Markdown vignettespage if it would be any helpful on the issue of sizing your Figures.
Aliyu Atiku Mustapha (11:27:20) (in thread): > Going through the document is exactly why I didn’t what to set my own figure dimensions. It stated clearly “BiocStyle comes with three predefined figure sizes.”
Emmanuel Taiwo devbird007 (11:57:16) (in thread): > Ohh, I get it now
Aliyu Atiku Mustapha (12:00:51) (in thread): > Yes. Suppose the dimensions for the 3 predefined sizes were stated, then I can use one of them and the figure wouldn’t be out of place. Hence the need to either leave it like that or use fig.width or fig.small to match the theme defaults
Dennis Ndubi (12:11:21): > Hello@Andres Wokatyi would like to know the progress of the review on my pr. I have made the last changes requested by@Paul Villafuerte. i would like to know if there are any other changes i may commit to my branch
Andres Wokaty (12:39:23) (in thread): > Let’s wait for Paul to confirm that it’s ready for me.
Dennis Ndubi (12:44:02) (in thread): > Okay..thank you
MaryJones (12:47:55): > @Andres WokatyI have deleted the .DStore file, so do I make new PR ?or is there is anything else
Andres Wokaty (12:51:07) (in thread): > So generally we try to avoid adding more dependencies (additional packages). Since the original showed a pdf, you can display the pdf; however, I think the last time someone tried this with html that it didn’t display properly. So what you can do is, make a .png of the pdf and display that with
Andres Wokaty (12:54:58) (in thread): > Commit your change to the same branch (as your current PR) and push that branch up so we can add to your PR.
Andres Wokaty (12:56:38) (in thread): > I’m glad you are still working on this. I know you were stressed out and I really want to help you better understand what we’re doing.
MaryJones (12:59:05) (in thread): > Ok, Thank you for assiting and understanding, I am grateful.
MaryJones (13:01:07) (in thread): > OK, I must make the change from my computer and push it like I did the first time but must not include the .DStore file ?
Andres Wokaty (13:06:23) (in thread): > Yes, that is correct. So you should do commands similar to > > git rm .DS_Store > git commit -m 'Remove .DS_Store' > git push origin chromBand-Rmd >
Aliyu Atiku Mustapha (13:12:23) (in thread): > okay, that I can do. I will still have to upload the png file with the Rmd file then, right? Thank you
Andres Wokaty (13:26:08) (in thread): > Yes, upload the .png and remove the .pdf
Aliyu Atiku Mustapha (13:38:43) (in thread): > just to inform you I had to make use of code chunk in able to name and caption the figure. >
Aliyu Atiku Mustapha (13:46:45) (in thread): > Thanks
MaryJones (13:47:23) (in thread): > Ok, thank you I will get to right away.
Paul Villafuerte (13:54:03) (in thread): > The last thing to do is to run these commands. YOu can check the Contribute documentation for more details. > * the tarball fromR CMD buildcontains theHTML(check with the following by substituting the package name and vignette nametar ztf package_name.tar.gz | grep 'doc/vignette_name')
Dennis Ndubi (13:55:22) (in thread): > Okay..lem them right now
MaryJones (14:03:04) (in thread): > Done !, please peruse, and advise further on how to update the PR., thank you..:pray:
Sonali Kumari (14:29:23) (in thread): > Hello,@Andres WokatyI hope I have completed all the tasks except the formatting of code chunks can you please guide me how to solve that problem ? as in should I need to reformat the code chunk in a consistent way for example in terms of indents etc.. can you please give me some guidance so that I can move forward in right direction, any sort of guidance will be appreciated.
Dennis Ndubi (14:47:21) (in thread): > @Andres Wokaty@Paul VillafuerteI have run the commands as shown below - File (PNG): image.png
{kind=link}
Dennis Ndubi (14:50:54) (in thread): > what should i do next@Paul Villafuerte@Andres Wokaty
Andres Wokaty (18:07:55) (in thread): > I appreciate you asking. Bioconductor has a coding style guide athttps://contributions.bioconductor.org/r-code.html#coding-style. Typically, 4 spaces are used for indents and I think I saw things not lining up well so that you know what’s an argument to a function, sohttps://style.tidyverse.org/functions.htmlmay help. - Attachment (contributions.bioconductor.org): Chapter 15 R code | Bioconductor Packages: Development, Maintenance, and Peer Review > Everyone has their own coding style and formats. There are however some best practice guidelines that Bioconductor reviewers will look for. can be a robust, fast and efficient programming language… - Attachment (style.tidyverse.org): 3 Functions | The tidyverse style guide > 3.1 Naming As well as following the general advice for object names, strive to use verbs for function names: # Good add_row() permute() # Bad row_adder() permutation() 3.2 Long lines There are…
2023-03-30
Dennis Ndubi (03:07:27): > Hello guys, hope you are fairing well. Just a random question, is it necessary to sign off commits for this PR?
Dennis Ndubi (03:46:40): > Hello@Andres Wokatyi have committed the changes requested to my pull. Kindly at your free time review and if more changes are needed, let me know. Thank you
Sonali Kumari (04:09:11) (in thread): > @Andres WokatyThank you for your help. I have already made the changes based on your feedback and I will review it again to ensure it meets the required standards. I have pushed the updated document and made the necessary changes. Once again, thank you for always guiding us in the right direction.
MaryJones (13:32:52): > @Andres WokatyHello everyone, Thank you for the review so as per**** the fig.caption and using an anchor doesn’t work: I have tried it before and I did that now here is how the display looks. And the cheat sheet doesn’t help cos you dont want me to use an image, you asked that I must use the generated image.. please kindly advice on what to do, thank you**.:pray: - File (PNG): Screen Shot 2023-03-30 at 5.48.19 PM.png
{kind=link}
Andres Wokaty (13:42:29) (in thread): > Please share the code on the PR so that I can give you better guidance.
MaryJones (13:44:23) (in thread): > here is the PR code:`{chr12ideogram, fig=TRUE, fig.align="center", echo=FALSE, prefix=FALSE, width=9, height=3} > build <- "hg19" ## or "hg18" > kp <- plotKaryotype(genome=build, chromosomes="chr12", main="Human chromosome12" ) > ## cyt2 <- getCytoband(build=build) > ## cyt2$gieStain <- "foo" > ## c12p12_idx <- intersect(grep("^q21", cyt2$name), > ## which(cyt2$chrom == "12")) > ## cyt2[c12p12_idx, "gieStain"] <- rep(c("gpos50", "gpos75"), > ## length=length(c12p12_idx)) > ## > ## > ## plotCytoband2(chromosome="12", build=build, cytoband=cyt2, outer=FALSE, ## From SNPchip > ## cex.axis=0.6, main="Human chromosome 12") > ## plotCytoband2(chromosome="12", build=build, cytoband=cyt2, outer=FALSE, > ## cex.axis=0.6, > ## main="Human chromosome 12") > >
Andres Wokaty (13:50:46) (in thread): > Where is the code you are modifying now?
Andres Wokaty (13:51:29) (in thread): > Can we please move this conversation to the PR?
Andres Wokaty (13:53:22) (in thread): > Let’s continue onhttps://github.com/Bioconductor/Category/pull/5#pullrequestreview-1365253550
MaryJones (13:57:51) (in thread): > it is there, already, I commented but I haven’t pushed the new code.
Andres Wokaty (13:58:20) (in thread): > Please paste the new code for that section in that discussion.
Andres Wokaty (14:06:14) (in thread): > I’ve replied in the PR so that we can document everything there.
Dennis Ndubi (14:09:02) (in thread): > hello@Andres Wokaty. this is to politely remind you i made the changes you requested on my PR. Kindly advice if there are other changes i should make to reach perfection
MaryJones (14:09:46) (in thread): > ok and the anchor produce lines, is that fine
Andres Wokaty (14:10:09) (in thread): > What do you mean by ‘produces lines’?
Andres Wokaty (14:11:03) (in thread): > Paul and I talked about it this morning as I had some confusion about if it was ready for me. So Paul needs to finish his review. I have one thing to add to your PR, though, which I will do shortly.
Andres Wokaty (14:11:51) (in thread): > I think I just guessed what it could mean. I’ll respond on the PR.
Dennis Ndubi (14:27:16) (in thread): > NP…mistakes are bound to happen for us to learn. i wil make the right changes right now
Dennis Ndubi (15:11:08) (in thread): > Hello, i have made the changes, and i thought it is better if I make the description item terms bold like in the first frame so that it will be easier to identify where the description begins to avoid any confusion with inline codes like in the second frame. I don’t know if that is a better idea and i need your advice. - File (PNG): image.png - File (PNG): image.png
{kind=link}
{kind=link}
Dennis Ndubi (16:00:48) (in thread): > @Andres WokatyKindly check my comment here
2023-03-31
MaryJones (01:05:41): > @Andres WokatyHello, all Kind see the PR, it updated and may please kindly review, I am the only left without no review even people who joined after me are already reviewed. Thank you
Saksham Gupta (06:37:42): > @Saksham Gupta has joined the channel
Andres Wokaty (09:17:36) (in thread): > We want to use the same styles for all vignettes so that the cues to the reader/user are all the same. Since these are arguments and not function names, it’s probably okay to change and I can see value in making them bolder and easy read, but maybe we shouldn’t mix the styles. So remove the backticks.
Dennis Ndubi (09:25:32) (in thread): > Okay..on it
khadijah (13:56:51): > @Andres Wokaty@Paul Villafuertei am having issues with the table and figure. how do i convert them. i have followed all the instructions in the docs - File (PNG): Screenshot 2023-03-31 at 18.55.37.png
{kind=link}
Paul Villafuerte (13:58:35) (in thread): > It looks like your missing a closing paren. Was kable in the converted code?
Dennis Ndubi (14:10:59): > Hello@Andres Wokaty@Paul Villafuertei have adjusted the changes to my PR
khadijah (14:13:44): > also for my figure, i am having issues with it - File (PNG): Screenshot 2023-03-31 at 19.12.42.png - File (PNG): Screenshot 2023-03-31 at 19.12.20.png
{kind=link}
{kind=link}
khadijah (14:14:26) (in thread): > the .rnw file doesnt come with a table code but its on the pdf
Andres Wokaty (14:31:20) (in thread): > I’m trying to understand what table you are referring to so that I can look at it in the sweave document first. Could you tell me where it starts in the PDF:https://bioconductor.org/packages/3.17/bioc/vignettes/DirichletMultinomial/inst/doc/DirichletMultinomial.pdfor in the Sweave code:https://github.com/mtmorgan/DirichletMultinomial/blob/master/vignettes/DirichletMultinomial.Rnw?
khadijah (14:38:41) (in thread): - File (PNG): Screenshot 2023-03-31 at 19.38.18.png
{kind=link}
khadijah (14:39:00) (in thread): > this table, apparently its not in the .rnw file
Andres Wokaty (14:43:57) (in thread): > This table, which isherein the .Rnw, is generated on line 248 in the code in your original image. I recommend troubleshooting why the code block isn’t producing the table.
Andres Wokaty (14:49:04) (in thread): > The plot should be generated by a code block. I don’t see it in the images you shared. In the .Rnw document, it is athttps://github.com/mtmorgan/DirichletMultinomial/blob/master/vignettes/DirichletMultinomial.Rnw#L78-L83.
khadijah (17:07:31) (in thread): > yes ma thats it
MaryJones (18:49:50): > @Andres WokatyHello Everyone can you give an update on the PR , I finished it since last night and can you please update on the review . thank you
2023-04-01
MaryJones (00:37:58): > @Andres WokatyPlease kindly check the pdf, I only convert from swerve to pdf, and edit so the document look like the pdf, I not know the arrangement, Kindly check the Pdf and swerve file to clarify, and kindly make up your mind.
MaryJones (00:41:07): > You instructed you want an anchor and for me to add an inline anchor according to the Html documents you gave us I need to add span tags else the lines will be formatted wrongly, so please , kindly clarify for sure that is what you want, as well as the YAML is on ly use d the render that work ,according to the YAML documentation you gave us , else if it indented wrongly it doesn’t compile, kindly clarify that too. Kindly list alll the changes at once so I do it once. Thank you.
MaryJones (01:23:29): > @Andres Wokaty****@refwas what I was up all night trying to fid solution , it never worked , I looked everywhere for the solution including this documentations you gave it never worked,I only used the one that worked.Can you just show me the code syntax . In my understanding the @ref code is a latex (.tex) syntax for referencing, Kindly show me how to use it in code. And I used html since the Rmd document can render Html.**
MaryJones (01:35:31): > @Andres WokatyThe YAML as you instructed see: - File (PNG): Screen Shot 2023-04-01 at 7.29.54 AM.png
{kind=link}
MaryJones (02:21:41): > @Andres WokatyPandoc converts : - File (PNG): Screen Shot 2023-04-01 at 8.08.24 AM.png
{kind=link}
MaryJones (03:40:19): > @MaryJones has left the channel
khadijah (09:28:44): > hi i made a pr already so i can record my contribution pending the time i get a review. here is the linkhttps://github.com/mtmorgan/DirichletMultinomial/pull/8 - Attachment: #8 Bioconductor/sweave2rmd: Conversion DirichletMultinomial.Rnw to Rmd > Bioconductor/sweave2rmd
> My name is Khadijah an Outreachy applicant. This is my second contribution I was assigned this issue on the project board, > > This pr is not that great, but i was hoping i would be able to record my contribution before the deadline and also get your feedback and corrections. Thank you for always being patient and helpful
> below is the > > HTML > > PDF > > cc @jwokaty
MaryJones (10:08:47): > @MaryJones has joined the channel
MaryJones (10:20:25): > Good day everyone: I wanted to quietly leave but on a second thought , no one deserves this type of treatments, I will share my experience, Since 27th I finished my project made pull request , ran R CMD and everyone see there is no error but since that 27th I get ask to be fix things even the ones that are on the Pdf, and I did up till today 1st April , no review !, and even when clearly branch are merging , i won’t get a merge either and I get told oh , I ran pandoc there is error which Is a lie !.. First, Mentors are supposed to build people not treat people like idiot, I feel like an idiot, is it the 10 times I have to delete this project and re-do it I want to think about, is the sitting up at night studying those documentations and other documentation to see that project work but I will get given a flimsy excuse to hide my hard work ? It hurts.
MaryJones (10:28:15): > Secondly , is it the fact that this mentor will go and tell another bald face lie that oh I can’t merge it cos she did a bad job and cost me the chance I needed to survive , I and my family.. No one should play on others emotion and intelligence , like it done to me today. Why the partiality ?, I will never understand and why I deserve this cruelty, I guess only the universe knows. I sat working , dedicated on this the entire time and with very little help ,get this done and I get played for a fool, till the time is gone , review I didn’t get and now I will loose the money I and my family so much need over a lie and partiality. Mentors are supposed to lift people up, not put people down. This mentor hurts me, I will never forget this ever, what this person did to me and I hope the universe remembers too..!
rohitsatyam102 (10:34:41) (in thread): > Hey I just used the PR link to register my contribution. I don’t think you should be worried about the PR request acceptance even making the final submit. Since the documents are evaluated for errors manually, I understand that this can be time taking task. And so I hope the outreachy mentors will take this into consideration.
MaryJones (10:39:30): > @MaryJones has left the channel
khadijah (10:40:38) (in thread): > yeah i understand. thank you
2023-04-03
Emmanuel Taiwo devbird007 (06:02:37): > Hi@Paul Villafuerte, I’ve been working on the note for a few days now and I am stumped. I explain more in my comment in the PRhere. > > Feedback would be appreciated.
rohitsatyam102 (08:49:32): > Hi Should I leave this box blank?<!channel> - File (PNG): image.png
{kind=link}
Andres Wokaty (08:51:42) (in thread): > Please refer the comments athttps://community-bioc.slack.com/archives/C03E7TWE2NT/p1679838635311959?thread_ts=1679838635.311959&cid=C03E7TWE2NTNT - Attachment: Attachment > when making the final application, there is a field that requires outreachy internship project timeline and it says to work with your mentor to provide a timeline of the work you plan to accomplish on the project and what tasks you will finish at each step. Could you @Andres Wokaty @Madelyn Carlson make some clarification here how one should go about it?
rohitsatyam102 (08:53:41) (in thread): > Thanks
2023-04-04
Busayo Samuel (she/her) (09:08:27): > I turned in my final application and wanted to take a moment to express my sincere gratitude to my amazing mentors and fellow contributors for their guidance and support throughout my Outreachy journey on the SweaveToRmd project. Your invaluable feedback, encouragement, and patience have truly made a difference in my learning and growth as an open source contributor and as a developer in general. Thank you for all for making this experience a memorable one. Cheers!!!
Andres Wokaty (11:17:37): > Thanks everyone for contributing. We’re still working through your reviews, which are taking time so you may receive a few items for feedback rather than a complete review.
Madelyn Carlson (13:07:40): > I want to take a moment to echo Jen, thank you everyone for contributing over the past month - you are all amazing and submitted wonderful work!
2023-04-05
Busayo Samuel (she/her) (10:51:57) (in thread): > @Andres Wokatyplease can more vignettes be added to the project board? I would love to continue contributing but all the vignettes have been currently assigned
Aliyu Atiku Mustapha (11:11:35) (in thread): > I second that!
Sonali Kumari (11:11:39) (in thread): > @Andres WokatyAlso can we work on current issues which are open in sweave2rmd project?
Andres Wokaty (11:19:05) (in thread): > I’ve cleared the ones we haven’t received PRs on so they’re now available. I would also accept PRs related tohttps://github.com/Bioconductor/sweave2rmd/issues/69andhttps://github.com/Bioconductor/sweave2rmd/issues/68.
Sonali Kumari (11:19:56) (in thread): > I would like to work on #69
Sonali Kumari (11:20:57) (in thread): > Should I comment on that issue ?
Andres Wokaty (11:23:12) (in thread): > I’ll assign you. I think I just want to make sure I don’t lose anything to review. Can we continue to usehttps://github.com/Bioconductor/sweave2rmd/issues/63to link our PRs?
Sonali Kumari (11:23:40) (in thread): > Ohk sure..
Andres Wokaty (11:24:00) (in thread): > Actually, I take that back@Sonali KumariCan you comment on #69.
Sonali Kumari (11:24:39) (in thread): > ohk I will do that.
Sonali Kumari (11:29:58) (in thread): > Also@Andres Wokatywe have all the PRs on the project board except this onehttps://github.com/mtmorgan/DirichletMultinomial/pull/8please attach this on the project board so that you will not forget about this PR.@khadijahis working on this PR. - Attachment: #8 Bioconductor/sweave2rmd: Conversion DirichletMultinomial.Rnw to Rmd > Bioconductor/sweave2rmd
> My name is Khadijah an Outreachy applicant. This is my second contribution I was assigned this issue on the project board, > > This pr is not that great, but i was hoping i would be able to record my contribution before the deadline and also get your feedback and corrections. Thank you for always being patient and helpful
> below is the > > HTML > > PDF > > cc @jwokaty
Busayo Samuel (she/her) (11:32:02) (in thread): > @Sonali KumariI think it’s already on the board
Sonali Kumari (11:32:34) (in thread): > Ohk I will check once again. Thank you@Busayo Samuel (she/her)I checked it was link of the .Rnw file
khadijah (11:34:14) (in thread): > @Andres Wokatywhen will I get a review on mine:pleading_face:
Andres Wokaty (11:50:54) (in thread): > We are trying to finish up the PRs with contributors who have not had a prior PR merged first. Please be patient.
Emmanuel Taiwo devbird007 (13:50:45) (in thread): > Understood, I currently just need@Paul Villafuerteto clarify on what he wanted me to include in the note in my PR.
Andres Wokaty (21:53:45) (in thread): > @Emmanuel Taiwo devbird007I think Paul just wanted you to describe the problem with summary in the initial PR comment and what you did, like you did with the figures. Sorry that the summary was so difficult. I’m going to see if I can find another example of this but I will take over the review.
2023-04-07
Emmanuel Taiwo devbird007 (13:52:30) (in thread): > Hi@Andres Wokaty@Paul Villafuerte, I ended up not needing the note since I was eventually able to fix the problem with the figures and make them align just as in the PDF, except those occurring in the middle of code chunk outputs of course. You can check out my latest comment and commitshere. - Attachment: Comment on #3 Convert RBGL.Rnw to RBGL.Rmd > Hello @villafup @jwokaty > > I know I was asked to make a note every time the pdf doesn’t match the knitted file.
> But instead I’ve spent the last few days rewriting the file to match the figures in the PDF file. > > The only ones I couldn’t match were those that appeared in the middle of a code output, and for those I placed the image immediately after the code output. > > Forgive me if I may be creating more work for you by making you go over it all over again, I will commit the new changes now. > > Do let me know what you would want me to do next.
2023-04-12
Dennis Ndubi (13:27:57): > Hello guys, you all went silent and its worrying. Hope you are all good
Sonali Kumari (13:45:49) (in thread): > Yes I am good… How about you?
Emmanuel Taiwo devbird007 (16:18:08) (in thread): > Yhup, good to hear from you Dennis.
Sonali Kumari (23:58:54): > @Andres WokatyI encountered an issue while documenting examples of code blocks I tried using 3 backticks (), triple backticks typically indicate that the enclosed content is meant to be interpreted as code, and it should be displayed as such when rendered or viewed. But This resulted in only half of the R code being interpeted, while the remaining part appeared as regular code chunks, which didn't look visually appealing. To address this, I tried using four backticks (````) instead of three, which partially resolved the issue, but the code chunk's name and other important information were not displayed, which could have been helpful for contributors. Finally, I discovered a better solution by using triple backticks () above the example and using double backticks (``) for the code block. This approach yielded the best results in terms of displaying examples of code blocks in a visually appealing manner. Nevertheless, I would like to inquire about the best approach for showcasing code block examples to users without actually executing the code. I have shared with you the results of code and knitted file after execution.
2023-04-13
Sonali Kumari (00:01:00) (in thread): > Output when we are using 4 backticks above example of code block. - File (PNG): 2023-04-13.png
{kind=link}
Sonali Kumari (00:02:47) (in thread): > Output when using 3 backticks above example. - File (PNG): 2023-04-13 (1).png
{kind=link}
Sonali Kumari (00:04:35) (in thread): > Best solution I was able to achieve, But I think there must be better approach to solve this.. Please guide me so that I can move forward. - File (PNG): 2023-04-13 (2).png
{kind=link}
Andres Wokaty (10:26:17) (in thread): > I looked for other examples of how to do this and saw that they use what looks like a hack on the bookdown site:https://bookdown.org/yihui/rmarkdown/r-package-vignette.html. You can see the code a the bottom ofhttps://github.com/rstudio/rmarkdown-book/blob/main/03-documents.Rmdfor the example > > ````markdown > `''````{r, fig.show='hold'} > plot(1:10) > plot(10:1) > > `> - Attachment (bookdown.org): 3.8 R package vignette | R Markdown: The Definitive Guide > The first official book authored by the core R Markdown developers that provides a comprehensive and accurate reference to the R Markdown ecosystem. With R Markdown, you can easily create reproducible data analysis reports, presentations, dashboards, interactive applications, books, dissertations, websites, and journal articles, while enjoying the simplicity of Markdown and the great power of R and other languages.
Sonali Kumari (11:01:56) (in thread): > Ohk thank you so much I will go through it and make the necessary required changes.
2023-04-18
Aliyu Atiku Mustapha (16:39:41): > @Andres WokatyGood afternoon. I just wanted to ask if it is still possible to still work on another vignette conversion, having had my last PR approved. > If yes, I would like to work on the ShortRead/vignettes/Overview.Rnw. > Thank you.
Andres Wokaty (16:40:21): > Hi, I know some of you have asked to work on additional vignettes; however, with the next release of Bioconductor coming next week, I will likely not be able to review any PRs until after the release. If you decide that you’d like to work on vignette anyway, I’m going to ask@Madelyn Carlson@Paul Villafuerte@Beryl Kanalito help with assignments. Thanks for your understanding and I really, really appreciate everyone’s contributions.
Sonali Kumari (23:14:42) (in thread): > @Andres WokatyI want to ask can I include the PR on issue #69 in my contribution to outreachy final application? I have not included it till now since I am not sure about it.
Andres Wokaty (23:29:41) (in thread): > Yes, you can add it if you are still able to update your application.
Sonali Kumari (23:36:49) (in thread): > We are not able to update our application but we can still record our contributions in a project we are interested in.
2023-04-19
Andres Wokaty (09:00:02) (in thread): > I see. Thanks for letting me know.
2023-04-27
rohitsatyam102 (13:29:45): > @Andres Wokaty@Madelyn CarlsonSome package vignettes do not have session info section that enlists versions of packages used while knitting them. I think this should be included in Sweave2Rmd best practices to have such a section at end of every vignette. Having attended BioC conferences, I have learned that it is good practice to do so (Just thinking out loud here).
Madelyn Carlson (14:02:08) (in thread): > Glad you posted! Good idea - tagging@Paul Villafuerteso that the and Jen can discuss @the next Sweave2Rmd check in.
rohitsatyam102 (14:06:57): > @Madelyn Carlson@Paul VillafuerteI would like to carry out Vignette conversion ofASpli/vignettes/ASpli.RnwKindly Assign this to me. > I would like to assign myself
Madelyn Carlson (14:14:24) (in thread): > I may be missing this vignette on the project board. Looks like w the re-release this package currently has a build error (https://bioconductor.org/packages/3.18/bioc/html/ASpli.html) & we would need to get approval from the maintainer. Unless this has already happened, perhaps claiming another vignette would require less steps? Project board:https://github.com/orgs/Bioconductor/projects/2/views/1?groupedBy%5BcolumnId%5D=Status - Attachment (Bioconductor): ASpli (development version) > Integrative pipeline for the analysis of alternative splicing using RNAseq.
rohitsatyam102 (14:17:36) (in thread): > Then kindly assign me any vignette that no one is working on
rohitsatyam102 (14:31:46) (in thread): > Okay I will go with GeneMeta
2023-04-28
rohitsatyam102 (11:27:49) (in thread): > @Madelyn CarlsonI have completed GeneMeta and raised a PR. I would like to start Genomic Alignments now
Madelyn Carlson (16:25:53) (in thread): > Apologies for missing your message yesterday. Amazing! Are you working on the summarizeOverlaps.Rnw vignette?
2023-04-29
rohitsatyam102 (01:37:05) (in thread): > Yes
2023-04-30
rohitsatyam102 (07:19:16) (in thread): > @Madelyn CarlsonGenomicAlignments vignette conversion completed and PR raised. Please update this on Project Board.
rohitsatyam102 (07:27:30) (in thread): > Taking ShortReadOverview.Rnwnow.
rohitsatyam102 (07:32:10): > @Andres Wokaty@Madelyn CarlsonAfter ShortRead, I would like to start with vignettes of DECIPHER Package. It is quite big however it is still in pre-triage and i couldn’t find its Github repo. So can you help?
Vince Carey (10:04:26) (in thread): > This package has been in Bioconductor for almost 12 years, so essentially predating the use of GitHub. There are several Rnw files in there. You could start working on them after cloninghttps://git.bioconductor.org/packages/DECIPHER– I don’t know the procedure for getting a GitHub repo set up for a pre-github package like this. You’ll have to use the contact-by-email approach athttps://github.com/Bioconductor/sweave2rmd/blob/bf163799ad54c605d9d90fa9a1d5110cebe5a5dd/vignettes/contribute.Rmd#LL76C6-L76C38
Vince Carey (10:05:21) (in thread): > It would be appropriate to ask the DECIPHER contributor whether they’ve considered managing their sources in GitHub. That would simplify matters.
2023-05-01
Andres Wokaty (09:46:38) (in thread): > @rohitsatyam102the DECIPHER maintainers should be contacted to see if they would accept a pull request but also before that as Vince mentioned, to see if they would consider managing their resources in Github. I do not recommend working on any of its vignettes until they’ve agreed to accept a PR as they may refuse or have preferences about which vignettes may be valuable enough to convert to .Rmd.
rohitsatyam102 (09:48:00) (in thread): > YES I have written to Erik for Decipher. But haven’t received any response. So I won’t start until I get a positive rejoinder from his side.
rohitsatyam102 (13:47:09): > How do I make table in the similar style as this? This table was made using latex - File (PNG): image.png
{kind=link}
rohitsatyam102 (13:47:50) (in thread): > Should the table look exactly same in Knitted HTML vignette?
Andres Wokaty (13:56:02) (in thread): > Can you share a link to the vignette?
rohitsatyam102 (14:06:12) (in thread): > https://www.bioconductor.org/packages/release/bioc/vignettes/ShortRead/inst/doc/Overview.pdf
Andres Wokaty (14:24:25) (in thread): > It might look a little different in R Markdown and that’s okay. Seehttps://bookdown.org/yihui/rmarkdown-cookbook/tables.htmlfor more on tables. - Attachment (bookdown.org): Chapter 10 Tables | R Markdown Cookbook > This book showcases short, practical examples of lesser-known tips and tricks to helps users get the most out of these tools. After reading this book, you will understand how R Markdown documents are transformed from plain text and how you may customize nearly every step of this processing. For example, you will learn how to dynamically create content from R code, reference code in other documents or chunks, control the formatting with customer templates, fine-tune how your code is processed, and incorporate multiple languages into your analysis.
rohitsatyam102 (15:35:48) (in thread): > Tried using Markdown format and span tag to create table. Works well when not using Biocstyle. However, when using biocstyle, the orange color specified using tag disapears.
Andres Wokaty (15:37:09) (in thread): > Don’t style to be orange, just use backticks to indicate these are functions or parameters
rohitsatyam102 (15:37:10) (in thread): - File (PNG): image.png
{kind=link}
rohitsatyam102 (15:38:47) (in thread): > I wanted to use it from aesthetic point of view. But okay
2023-05-02
rohitsatyam102 (05:06:42): > @Andres WokatyPlease update the Project Board for the following: PR request generated for ShortReadOverview.Rnw, Genomic AlignmentssummarizeOverlaps``.rnwand geneplottervisualize.Rnw.
Andres Wokaty (10:25:33) (in thread): > What should their status be? And if there are PRs, could you provide the links?
2023-05-03
rohitsatyam102 (03:42:36) (in thread): > Oh yes I can provide the links. I also tagged you in those
rohitsatyam102 (03:43:15) (in thread): > ShortRead:https://github.com/Bioconductor/ShortRead/pull/14 - Attachment: #14 Converted .Rnw to .Rmd and edited DESCRIPTION File > Asking for review from @Bioconductor/sweave2rmd @jwokaty @mcarlsn for vignette Overview.Rnw conversion. Below attached is the HTML output of the vignette.
> Overview.zip
rohitsatyam102 (03:43:40) (in thread): > GenomicAlignments:https://github.com/Bioconductor/GenomicAlignments/pull/28 - Attachment: #28 Converted .Rnw to .Rmd and edited DESCRIPTION file > Asking for review from @Bioconductor/sweave2rmd @jwokaty @mcarlsn for vignette summarizeOverlaps.Rnw conversion. Below attached is the HTML output of the vignette.
> summarizeOverlaps.zip
rohitsatyam102 (03:44:02) (in thread): > GeneMeta:https://github.com/Bioconductor/GeneMeta/pull/2 - Attachment: #2 GeneMeta rmd > Asking for review from @Bioconductor/sweave2rmd @jwokaty @mcarlsn. Below attached is the HTML output of the vignette.
> GeneMeta.zip
rohitsatyam102 (03:44:49) (in thread): > geneplotter(two Vignettes in same PR) :https://github.com/Bioconductor/geneplotter/pull/2 - Attachment: #2 Convert byChroms.Rnw to byChroms.Rmd > Asking for review from @Bioconductor/sweave2rmd @jwokaty. Below attached is the HTML output of the vignette.
> byChroms.zip
rohitsatyam102 (03:45:58) (in thread): > Their status is stilltodoso maybe you can change it and also assign the name against the contributor.
Sonali Kumari (08:25:27) (in thread): > @Andres WokatyI want to work on this issue #68 (https://github.com/Bioconductor/sweave2rmd/issues/68) . I have noticed this issue while working on #69 I have mentioned it here-(https://github.com/Bioconductor/sweave2rmd/pull/70#issuecomment-1513491062) and I think I can solve this issue and I will help you solve this issue as a point of view of contributor I will make sure to add EXAMPLES nicely so that new contributors won’t face issues which I have faced in my initial conversion so that it will be simplified for the future contributors. Should I comment on that issue ? - Attachment: #68 Update Contribution to clarify manual edits - Attachment: Comment on #70 Update checklist
Andres Wokaty (09:28:23) (in thread): > Yes, please. I plan to review your other PR this week.
Sonali Kumari (09:31:09) (in thread): > Thank you Jen:blush:.
Andres Wokaty (09:43:39) (in thread): > I’ve updated the board. Let’s focus on finishing the reviews and getting them merged before doing any additional ones. I think the closest to merging ishttps://github.com/Bioconductor/geneplotter/pull/2. I’ll review this week. - Attachment: #2 Convert byChroms.Rnw to byChroms.Rmd > Asking for review from @Bioconductor/sweave2rmd @jwokaty. Below attached is the HTML output of the vignette.
> byChroms.zip
rohitsatyam102 (09:44:02) (in thread): > Yes please
2023-05-04
rohitsatyam102 (08:02:47): > @Andres Wokatythe request for Decipher Package was declined. Please update that on Trello Board - File (JPEG): Screenshot_2023-05-04-17-31-39-63_e307a3f9df9f380ebaf106e1dc980bb6.jpg
{kind=link}
rohitsatyam102 (09:13:06) (in thread): > Also it is high time there is a rationale section on Sweave2Rmd page so that people can understand the underlying cause behind this project. Erik asked me this question. - File (JPEG): Screenshot_2023-05-04-18-41-28-60_e307a3f9df9f380ebaf106e1dc980bb6.jpg
{kind=link}
Andres Wokaty (09:14:30) (in thread): > If you would like to work on a PR for that, please do
Madelyn Carlson (10:13:22) (in thread): > Thanks so much for your work on this,@rohitsatyam102! FYI I officially transitioned out of my role with Waldron Lab & Sweave2Rmd recently so my response times will be slower. Happy to answer any questions but for quicker responses and PR reviews, you can tag Paul or Jen.
rohitsatyam102 (10:19:41) (in thread): > Sad to hear that@Madelyn Carlson. You will be missed. Thanks for letting us know. Best of luck for the new role elsewhere wherever you join.
Madelyn Carlson (10:20:19) (in thread): > Thank you so much,@rohitsatyam102! Really appreciate your message
rohitsatyam102 (10:24:02) (in thread): > Will do
Dennis Ndubi (12:40:12): > Congratulations@Sonali Kumari
Dennis Ndubi (12:42:24): > For those of us whom we didnt manage this round, there is a next round and dont give up
Busayo Samuel (she/her) (12:49:24) (in thread): > :tada:Congratulations Sonali!!!
sophy (13:01:44) (in thread): > Congratulations Sonali:tada:
sophy (13:02:04): > @sophy has left the channel
Sonali Kumari (13:08:38) (in thread): > Thank you so much@Dennis Ndubi@Busayo Samuel (she/her)@sophy
Oluwabukola Bamigbade (14:38:28): > @Oluwabukola Bamigbade has left the channel
2023-05-05
Aliyu Atiku Mustapha (15:35:30): > @Aliyu Atiku Mustapha has left the channel
2023-05-06
rohitsatyam102 (14:39:49) (in thread): > Congrats@Sonali Kumari
rohitsatyam102 (14:40:14) (in thread): > Yes I will apply again.
rohitsatyam102 (14:45:13): > @Andres Wokaty@Paul Villafuerte@Beryl KanaliI have seen in many PRs that even the spaces are being pointed out in the YAML section even when the Vignette is getting produced without any error. Why is it important (I understand spaces are important in main text).
rohitsatyam102 (14:57:37) (in thread): > @Andres Wokaty@Paul Villafuerte@Beryl Kanaliwas this discussed?
Sonali Kumari (15:26:55) (in thread): > Thank you@rohitsatyam102
Andres Wokaty (18:36:32) (in thread): > Not yet. I have been wrapped up in release activities but we should discuss at the next meeting. These are generally Thursday morning ET if you’d like to attend. However, this probably needs some discussion with the core as generally sweave2rmd tries not to alter the vignette unless there is good reason, such as a broken plot.
Andres Wokaty (18:48:39) (in thread): > sweave2rmd strives to make older vignettes maintainable but just because it is converted to .Rmd doesn’t make it automatically easily to maintain. It should be readable and we can accomplish this through some style guides, which is what is currently missing in the project’s documentation. You learn it somewhat through the review process but it should be formalized with a written guide. It’s the same with any code: you can write code that runs but is hard to modify/maintain because you or the next person who needs to edit it can’t read it. This is why there are a lot of comments and requests to fix formatting.
2023-05-09
Rukky (02:41:34): > @Rukky has left the channel
2023-05-13
rohitsatyam102 (15:11:19): > Hi@Andres Wokaty. I think it will take u some time to review all the PRs that I raised. Meanwhile could u please assign me another vignette? I want to keep practicing
Andres Wokaty (17:29:06) (in thread): > Instead of doing another vignette, could I ask you to review your PRs using the checklist athttps://bioconductor.github.io/sweave2rmd/articles/contribute.html#review-a-pull-request? I am the person primarily doing reviews at this point, which is why it takes time as I am also doing other tasks. Usually, after someone has done a few PRs, I suggest they start doing reviews too because it also improves the quality of their PRs. I am going to review your GeneMeta PR next so maybe you can review ShortReads? - Attachment (bioconductor.github.io): Contribute > Learn how to contribute to our project.
2023-05-14
rohitsatyam102 (14:05:32) (in thread): > Okay. Sounds good to me. I RECEIVED your emails. Will do the needful
Sonali Kumari (15:22:29) (in thread): > I will work on this issue-[https://github.com/Bioconductor/sweave2rmd/issues/68] after my exams ends that is from 17th may but it is not assigned to me till now is it ohk to work on that issue? and also@Andres WokatyI want to contact maintainers of affy package for sweave2rmd conversion request should I open a issue on their repository or send mail to the maintainer directly? I will send them mail as written in contribute document additionally I will provide them information about why .Rmd is preferred over .Rnw? - Attachment: #68 Update Contribution to clarify manual edits > The current contribution document gives the impression that all that is needed is to run pandoc without errors to generate a successful .Rmd, but this is misleading. In particular, there should be a section to expand on what needs manual correction in steps current 13 and 16. This can include examples of code. > > Additionally, there should be another step that considers further how to make code easier to maintain, for example, by using Reflow Comment to break up long lines.
Sonali Kumari (15:31:36) (in thread): > I want to start early because this project is really interesting to me. It is my first live project, and I truly enjoy working on it:party_parrot:
2023-05-15
Andres Wokaty (09:10:52) (in thread): > I’ve assigned you issue 68; however, I don’t think we should start on affy yet because it has an error on the build report. We usually want to work on packages that are passing all stages so that we know if our code is the problem. I’m going to think about what packages we should try to work on and if we should change the process a little this week.
Sonali Kumari (09:19:05) (in thread): > ohk@Andres Wokaty. If you want me to work on any other packages please let me know , as I am available to start working from the 17th. Additionally, I was wondering if there is a method or process we can utilize to identify any potential build errors in the packages.
Andres Wokaty (09:20:45) (in thread): > I usually look at the software build report athttps://bioconductor.org/checkResults/3.18/bioc-LATEST/.
Andres Wokaty (09:21:24) (in thread): > Do you mean when you are working on a package?
Sonali Kumari (09:23:01) (in thread): > No. I was just asking for my knowledge so that from next time I will check if needed
Sonali Kumari (09:26:46) (in thread): > I also understand by your msg that in our project we work on a package that usually does not have build error so I think we could check before contacting the maintainer if I am not wrong.
Andres Wokaty (09:29:10) (in thread): > Yes, that’s right
Sonali Kumari (09:32:12) (in thread): > Ohk thank you.
Andres Wokaty (09:32:29) (in thread): > Good luck on your exams!
Sonali Kumari (09:33:45) (in thread): > Yes thank you. I have my last exam tomorrow and I am already excited for my internship hahahahah:party_parrot:
Andres Wokaty (11:58:15) (in thread): > Next week, if you have time and are interested, Bioconductor is participating in GTN Smorgasbord athttps://gallantries.github.io/video-library/modules/bioconductor. These are free online classes at your own pace. This is the first time Bioconductor is participating. Some of the material is really over my head but in the past, I’ve suggested parts of The Bioconductor Project course. The first few sections provide a good overview of the project and things like how to install packages. - Attachment (gallantries.github.io): Bioconductor R analyses
Sonali Kumari (12:26:30) (in thread): > I will definitely go through these. I am sure these are helpful for me. Thank you!
2023-05-18
Oluwafemi Oyedele (05:54:02): > @Oluwafemi Oyedele has joined the channel
2023-05-26
Wossen Mengesha (07:42:15): > @Wossen Mengesha has joined the channel
2023-06-08
Janani Ravi (20:58:12): > @Janani Ravi has left the channel
2023-06-11
rohitsatyam102 (13:49:52) (in thread): > I found the figure in this tweet could be used to show how various file formats can branch out from .RMD files:https://twitter.com/maximaDataSci_R/status/1667423109366906880?t=0f5Um3A3CUBFM0aP7FMDwQ&s=19 - Attachment (Twitter): MÁXIMA FORMACIÓN Data Science_R on Twitter > R Markdown convierte tus análisis de datos en documentos, informes, presentaciones y tableros de mando de alta calidad.¡Ya no más copy & paste! Te preguntarás cómo has podido vivir sin ella. https://t.co/eLLJRtae8L #master #cursos #formacion #empleo #rstats
2023-06-12
Andres Wokaty (12:00:16) (in thread): > This would be good to add to the documentation for sweave2rmd.
Andres Wokaty (12:02:13) (in thread): > Also addingsessionInfowas discussed at a meeting, and it’s good to add it at the end of vignettes. If it’s added to contributed package’s vignette, it should be mentioned so that the maintainer knows it was added.
2023-06-13
Dennis Ndubi (11:57:06): > Hello, i would like to work on any availabe vignette. Can be assigened one please?
Sonali Kumari (12:02:55) (in thread): > Yes thanks for asking, I will assign you Category/vignettes/ChromBand.Rnw from theproject board
Sonali Kumari (12:03:27) (in thread): > After completing the conversion can you please send the link of your PR in this thread so that we will update the PR in the board?
Dennis Ndubi (12:22:04) (in thread): > Okay., no problem
2023-06-16
Shivang Bhanushali (08:55:36): > @Shivang Bhanushali has joined the channel
Dennis Ndubi (17:10:55): > @Andres Wokaty@Sonali KumariEncountered this while do the conversion with the pandoc command . The second frame is line 305 where that error arises. I don’t understand where since I see there is an opening and closing for the section. - File (PNG): image.png - File (PNG): image.png
{kind=link}
{kind=link}
Andres Wokaty (17:36:28) (in thread): > Sometimes pandoc has difficulty converting chunks like this. I am not sure why. I recommend removing the chunk (figure) then running pandoc on the .Rnw. Then you can manually insert the chunk back into the .Rmd but in the “R Markdown” format.
2023-06-17
Dennis Ndubi (04:44:17) (in thread): > The error still persists, Kindly directly nme what to do
Sonali Kumari (04:56:45) (in thread): > Hello Dennis I am familier with this problem. The advice given by jen is the way to solve the problem In your case, I think there is a problem in /end {figure} I recommend removing all the code chunks which include this /end {figure} from .Rnw file and then try run pandoc code in .Rnw file
Sonali Kumari (04:57:51) (in thread): > After successfully running the pandoc code please add those figure in new .Rmd file in “R Markdown” format.
Sonali Kumari (04:58:55) (in thread): > If you still have any questions and if the error still persists please feel free to ask we will try to solve your problem together in a quick slack huddle.
Dennis Ndubi (05:10:59) (in thread): > Sure …ill will dp that
2023-06-22
Levi Waldron (08:51:38): > Hi<!channel>(especially@Andres Wokaty) - I think I have a working template repo for automatical default conversion of the Rnw files of a Bioconductor package to Rmd using GitHub Actions. Would someone give it a try and see if it works for you?https://github.com/lwaldron/sweave2rmd_conversiontemplate
Sonali Kumari (16:10:47) (in thread): > Thanks for this amazing opportunity, I will work on this and share the results with you soon.
2023-06-23
Levi Waldron (10:29:48) (in thread): > Did this rationale get updated? I’m just catching up a bit, but I would say we hope to eventually deprecate Sweave because it does not support BiocStyle, is not compatible with mobile devices or dark backgrounds, and produces PDF vignettes that are difficult for anyone with poor eyesight to read. I would’ve told Erik that he may not have someone to do it for him later:wink:
Andres Wokaty (10:33:43) (in thread): > I made an issue for this, which is still open. If@rohitsatyam102would like to work on it, it’s still available.
rohitsatyam102 (12:26:56) (in thread): > Since there is no GitHub repo, I wrote an email to Erik. But now that Levi has mentioned that you guys are planning to depreciate the Sweave format, it might change his mind. I am sorry I am in a bit of a crisis right now with my mother’s health and busy in taking care of her. But I will write an email anyway and would love to contribute to this package since the number of vignettes are many and it would be a good learning opportunity.
Levi Waldron (12:28:56) (in thread): > Don’tworry@rohitsatyam102! If you send me his emailI’llfollow up.It’stoo early to say we will deprecate Rnw for sure but that is the goal.
rohitsatyam102 (12:30:08) (in thread): > Thanks Levi. Here it is:eswright@pitt.edu
2023-06-24
Sonali Kumari (11:08:19) (in thread): > Hello@Levi Waldron, I wanted to share with you that I tried using the annotate package of Bioconductor, and it worked well! It converted all the .Rnw files to .Rmd files and found it to be very useful overall. There were a few issues with the conversions, but I’m interested in working on them. Thank you for introducing us to this idea.
Sonali Kumari (11:27:02) (in thread): > GitHub action takes about 22 minutes to complete for this conversion.
Levi Waldron (14:17:45) (in thread): > Great, Sonali! The conversion is done byhttps://github.com/LiNk-NY/Rnw2Rmdwhich we can make issues or pull requests on for conversion issues.
2023-06-25
Sonali Kumari (05:02:54) (in thread): > Thank you.
2023-07-12
Sonali Kumari (10:41:42): > @Andres WokatyThis is the branch where I pushed my current changes regarding the conversion of GWASTolls Datacleaning.Rmd -https://github.com/sonali8434/GWASTools/tree/DataCleaning-RmdThis file is too long and we are thinking that if converting such long file is really worth or not?
2023-07-13
Sonali Kumari (10:19:22): > @Andres WokatyAfter reading the comment of alexander in fastseg PR -https://github.com/alexg9010/fastseg/pull/1#issuecomment-1634301696I think we should move the scope of document above toc but it will add there as a abstract so the heading will not be there . Is it fine?
Sonali Kumari (10:25:36) (in thread): > And also they asked me add myself in the description file so, I will not add myself on the .Rmd?
Andres Wokaty (10:37:26) (in thread): > I think it’s fine since it is their suggestion. I think you could possibly add the title since it appears that the section will still render R Markdown; however, it may still showAbstractthere. You could maybe a screenshot or use Rpub to share what it would look like to help them decide. > > As for adding yourself as a contributor, it does sound like they just want you to be added in the DESCRIPTION file (and don’t mention the .Rmd). I see that in your discussion with them that you mentioned both places where we typically add contributors.
Sonali Kumari (10:39:00) (in thread): > Ok.. I will fix the abstract and add myself as a contributor in the description file.
Andres Wokaty (11:01:46) (in thread): > I have a few thoughts about this. As I mentioned at our meeting, the maintainer should be involved in discussions about whether they think it’s worth converting the vignette because documentation gets outdated. However, if users are still using this vignette then there’s value in converting it. The maintainer probably has the best sense of this and this is a discussion we should have with them when we first approach them about conversions.@Levi WaldronI wanted to ask your advice about discussing with maintainers whether to convert a vignette that might be possibly outdated. In this example, the DataCleaning.Rnw has an old URL for a project. I know just because something is old it could still be widely used, but it’s possible that there other methods that are preferred these days.
Sonali Kumari (11:30:35) (in thread): > @Andres Wokatythis is the best I was able to achieve but I am happy this is good -https://rpubs.com/Sonali8434/1062430
Sonali Kumari (11:31:59) (in thread): > I was also able to add blank line( carriage return) in abstract thanks to@Paul VillafuerteI have taken help from the issue he created for the same.
Andres Wokaty (11:33:05) (in thread): > Great. Please share with maintainer and remember to remove the other other section in the document below the ToC:wink:
Sonali Kumari (11:33:41) (in thread): > yes Thanks for reminding me to remove the extra part:rolling_on_the_floor_laughing:
Sonali Kumari (15:20:49): > Hello@Andres WokatyI am having issues with the loom I have recorded the whole video:smiling_face_with_tear:But everytime sound is not being recorded I have given the camera and mic permission then also I am having issues. -https://www.loom.com/share/192d5fab308a4fd3a64b219968f791e6
Andres Wokaty (15:26:02) (in thread): > I’m sorry, that’s really frustrating!
Sonali Kumari (15:26:41) (in thread): > What to do?
Andres Wokaty (15:28:13) (in thread): > Maybe we should look for other software? It’s too bad because the recording looks good!
Andres Wokaty (15:31:44) (in thread): > I think for example zoom has a feature to record.
Sonali Kumari (15:32:52) (in thread): > Let me try too troubleshoot and try once more .. Then I will look for another options.. Hope this works:melting_face:
2023-07-14
Levi Waldron (17:38:22): > @Andres WokatyI feel like you asked a question somewhere that Ican’tfind because I can no longer find anything on Slack if I don’t get to it immediately:sweat_smile:. About improvements to outdated documentation or broken links?
2023-07-17
Sonali Kumari (06:35:27): > Hello@Andres WokatyI have made a PR for the changed github actions and .Readme file for the Levi’s package template please review my PR I have made the changes to readme file -https://github.com/waldronlab/sweave2rmd_conversiontemplate/pull/2
Andres Wokaty (09:40:54) (in thread): > I have a general question about talking with maintainers about what vignettes should be converted because they can be outdated. For example, we have a long vignette that@Sonali Kumaristarted working onhttps://github.com/sonali8434/GWASTools/tree/DataCleaning-Rmd, which describes a process for data cleaning from around 2010 and a project that may no longer exists at the URL described in the vignette. This could still be the way to do things, so it would be important to convert it; however, it could also be an outdated method at this point. > > I think we should go back to the maintainers and talk to them, although I’m not sure how exactly, because it’s a big vignette that would take a lot of time and effort.
Andres Wokaty (18:27:01) (in thread): > I’ll wait for a response from Levi before finishing the review.
2023-07-18
Sonali Kumari (03:08:31) (in thread): > Ok
2023-07-20
Sonali Kumari (14:04:58): > Hello@Andres WokatyWolfgang Huber replied to my mail, but I need help understanding what he was asking. Can you please explain that to me? I am still getting familiar with some of the words written in that mail.
Andres Wokaty (15:25:21) (in thread): > Wolfgang agreed to accept conversions, but suggested that rather than using R Markdown to useQuartoinstead. Quarto is a new format that can be used with different languages, like R and Python. I think the format is similar to R Markdown. This might be a good small project to assess what changes we might make to allow this as possible alternative to .Rmd. - Attachment (quarto.org): Quarto > An open source technical publishing system for creating beautiful articles, websites, blogs, books, slides, and more. Supports Python, R, Julia, and JavaScript.
Sonali Kumari (15:30:06) (in thread): > That Makes sense, thank you.
2023-07-25
Sonali Kumari (12:50:10): > Hello@Andres WokatyI am using this command(from = "us", to = "mnt/STORE1/bighome/skumari/KEGGgraph/vignettes/KEGGgraph.Rmd", validate = FALSE)in the terminal, but I am getting error msg > > Command 'from' not found, but can be installed with: > apt install mailutils > Please ask your administrator. >
Andres Wokaty (12:51:09): > Can you share a screenshot?
Sonali Kumari (12:52:22): - File (PNG): RStudio Server and 4 more pages - Personal - Microsoft Edge 25-07-2023 22_21_36.png
{kind=link}
Andres Wokaty (12:57:35): > So you want to change from the terminal to the R console. Is this for Rnw2Rmd? You can set the path in the R console withsetwd()
Andres Wokaty (12:58:06): > Or you can set the full path for both the from and to parts.
Sonali Kumari (13:03:18) (in thread): > its now giving this.. - File (PNG): RStudio Server and 4 more pages - Personal - Microsoft Edge 25-07-2023 22_32_18.png
{kind=link}
Sonali Kumari (13:06:39) (in thread): > ohhh I am giving the wrong path sorry
Andres Wokaty (13:06:45) (in thread): > Rnw2Rmd(from="/mnt/STORE1/bighome/skumari/KEGGgraph/vignettes/KEGGgraph.Rnw", to="/mnt/STORE1/bighome/skumari/KEGGgraph/vignettes/KEGGgraph.Rmd", validate=FALSE)
Andres Wokaty (13:06:48) (in thread): > yes!
Andres Wokaty (13:07:28) (in thread): > you also need the beginning slash in front of/mnt
Sonali Kumari (13:08:05) (in thread): > ok I will try and share the results with you.
Sonali Kumari (13:10:11) (in thread): > still having the same issue - File (PNG): RStudio Server and 4 more pages - Personal - Microsoft Edge 25-07-2023 22_39_27.png
{kind=link}
Andres Wokaty (13:11:54) (in thread): > Can we do a quick huddle?
Sonali Kumari (13:12:20) (in thread): > yes
Sonali Kumari (13:19:45) (in thread): > @Andres WokatyThank you so much. I found this method super easy.
Andres Wokaty (13:20:57) (in thread): > Great!
Andres Wokaty (13:21:17) (in thread): > We can use it going forward.
Sonali Kumari (13:38:46): > @Andres WokatyHow about this?https://github.com/Bioconductor/sweave2rmd/issues/85#issuecomment-1650261333
Andres Wokaty (13:43:23) (in thread): > You can close it. Thank you!
2023-07-26
Sonali Kumari (14:22:28): > Hello@Andres Wokaty, I have done this conversion -https://github.com/kasperdanielhansen/Rgraphviz/pull/21recently. Please review it whenever you have some free time. I have updated it on a board.
2023-07-27
rohitsatyam102 (03:23:16): > Hi@Sonali Kumari@Andres Wokatyif you guys have any vignette conversion task that you could assign me. Several of my PRs were merged recently and I wish to contribute to more vignettes.
rohitsatyam102 (03:24:39) (in thread): > Hi wanna follow up on this if Erik is on board for vignette conversion?
Levi Waldron (03:25:24) (in thread): > Yes, thanks for the reminder:sweat_smile:
rohitsatyam102 (03:30:23) (in thread): > Now that I am back and working, I can write an email explaining the possibility of complete migration to .RMD and possible depreciation of .Rnw format if you want
rohitsatyam102 (03:32:06) (in thread): > If there are RNAseq or scRNAseq related vignettes or visualization related please let me know. Those would be most beneficial for me
Levi Waldron (03:51:34) (in thread): > Don’tworry,I’llwrite it
Andres Wokaty (09:42:02) (in thread): > I’m just including@Sonali Kumariin the conversation since she’s also doing a lot of communications with maintainers.
Sonali Kumari (09:47:46) (in thread): > I have left few comments on GeneMeta please do the needful corrections then it will be ready for jen to review, also I have left one comment regarding date on shortread after that I think shortread is ready to be merged.
Sonali Kumari (09:48:42) (in thread): > We will surely assign you a vignette after the discussion with Jen.
2023-07-29
Sonali Kumari (10:14:31) (in thread): > @rohitsatyam102we have discussed and realized that we don’t have any visualization-related vignettes on the board currently. You can search for your choice of vignette, and then you may ask the maintainer. We have updated thecontributiondocument regarding communication with maintainers. You can follow the same approach to contacting them. > Thank you. - Attachment (bioconductor.github.io): Contribute > Learn how to contribute to our project.
rohitsatyam102 (10:38:35) (in thread): > Okay Sonali. Will do
2023-08-07
KritikaVerma (09:53:08): > Hello! > I wanted to contribute to bioconductor community. Is there a vignette that I can be assigned?
Sonali Kumari (12:06:39) (in thread): > Yes, sure, I will assign you to annotate/vignettes/chromLoc.Rnw, can you please give me your GitHub user id
KritikaVerma (12:20:13) (in thread): > Thank you! Here is my GitHub ishttps://github.com/SunSummoner
Sonali Kumari (13:02:27) (in thread): > I have assigned you that you can work on this conversion. Please send the link to your PR in this thread after doing the conversion.
KritikaVerma (13:20:00) (in thread): > OK, thanks!
2023-08-14
Sonali Kumari (06:37:53): > @Andres WokatyI am converting one file in which there are sections where we refer to pages, but we don’t have pages in HTML. What to do in such cases?
Sonali Kumari (09:31:00) (in thread): > You can see my changes here in this branch-https://github.com/sonali8434/KEGGgraph/tree/KEGGgraph-Rmd
Sonali Kumari (09:32:24) (in thread): > at line 231 we have pageref{}
Andres Wokaty (10:32:07) (in thread): > I think this happens often. I recommend that bringing this to the maintainer’s attention in the PR and providing a suggestion, such using the word ‘section’ in place of ‘page’ and a link to the appropriate section in place of thepageref{}.
KritikaVerma (14:24:08) (in thread): > @Sonali KumariI am bit confused is there a read me or instructions for converting the vignettes?
2023-08-15
Sonali Kumari (05:13:47) (in thread): > please refer this contribute document -https://bioconductor.github.io/sweave2rmd/articles/contribute.html - Attachment (bioconductor.github.io): Contribute > Learn how to contribute to our project.
2023-08-16
KritikaVerma (07:27:40) (in thread): > Thank you!
2023-08-22
Reece (10:37:58): > heylo@Sonali Kumari@Andres WokatyIs there any vignette that i can be assigned ? would want to contribute and learn from the community
2023-08-24
Sonali Kumari (10:17:43) (in thread): > Thanks for asking. I will assign you this - annotate/vignettes/query.Rnw Please feel free to contact any of the community members for help. Preferably, you can tag me or Jen for doubts related to conversion.
Sonali Kumari (10:20:43) (in thread): > Please send your GitHub user ID to this thread? Also, after making a PR, please send the PR link in this thread so we can review your PR soon.
Reece (10:25:31) (in thread): > https://github.com/Reece11
2023-09-13
shaimaa Elsafoury (09:51:15): > @shaimaa Elsafoury has joined the channel
2023-09-14
KritikaVerma (15:19:12): > Hello everyone! > I had a problem while usingknit. This was theyamlfile:---``title: "An Introduction to the GenomicAlignments Package"``author: "Herv\'{e"``date: "format(Sys.time(), ‘%B %d, %Y’)"``output:`` BiocStyle::html_document``vignette: >`` %\VignetteIndexEntry{ An Introduction to the GenomicAlignments Package }`` %\VignetteEngine{knitr::rmarkdown}`` %\VignetteEncoding{UTF-8}``---This is the error I am getting: > > Error in yaml::yaml.load(..., eval.expr = TRUE) : > Scanner error: while parsing a quoted scalar at line 2, column 9 found unknown escape character at line 2, column 14 > Calls: <Anonymous> ... parse_yaml_front_matter -> yaml_load -> <Anonymous> > Execution halted >
Marcel Ramos Pérez (16:13:41) (in thread): > It looks like the author should beHervé?
Marcel Ramos Pérez (16:14:14) (in thread): > Hervé Pagès
KritikaVerma (22:40:32) (in thread): > That makes sense! Thank you!
2023-09-17
KritikaVerma (02:01:26) (in thread): > After converting into the Rmd do I have to remove the Rnw vignette from the repository?
Marcel Ramos Pérez (03:45:29) (in thread): > Yes
2023-09-20
KritikaVerma (03:12:48): > > processing file: chromLOC.Rmd > |........................ | 40% [buildCL] > Quitting from lines 48-51 [buildCL] (chromLOC.Rmd) > Error: > ! getAnnMap: package hgu95av2 not available > Backtrace: > 1. annotate::buildChromLocation("hgu95av2") > 2. annotate::getAnnMap("CHRLOC", dataPkg) > 3. base::tryCatch(...) > 4. base (local) tryCatchList(expr, classes, parentenv, handlers) > 5. base (local) tryCatchOne(expr, names, parentenv, handlers[[1L]]) > 6. value[[3L]](cond) > > Execution halted > > While trying to knit a vignette I was getting this error.
Andres Wokaty (09:52:59) (in thread): > Reading that error, it sayspackage hgu95av2 not available. I wouldBiocManager::install("hgu95av2")and see if that helps move it further along.
KritikaVerma (14:39:30) (in thread): > @Andres WokatyThank you! It worked.
2023-09-25
KritikaVerma (22:34:42) (in thread): > Hiihttps://github.com/Bioconductor/annotate/pull/3This is the link to the PR - Attachment: #3 Convert the cromLOC vignette and added the Rmd file. > I converted the vignette assigned to me and removed to old file. Please review the PR.
2023-09-26
Sonali Kumari (15:01:46) (in thread): > Thank you for your PR we will add this on board and review it as soon as possible.
2023-09-27
KritikaVerma (23:30:00) (in thread): > This the new PRhttps://github.com/Bioconductor/annotate/pull/4 - Attachment: #4 Convert file to chroRmd > ☑︎ the .Rmd file knits to HTML > ☑︎ R CMD build runs without errors or timeouts > ☑︎ the tarball from R CMD build contains the HTML (check with the
> following by substituting the package name and vignette name tar ztf package_name.tar.gz | grep 'doc/vignette_name') > ☑︎ the .Rnw file has been removed > • In the DESCRIPTION file > ☑︎ BiocStyle and knitr are listed in Suggests > ☑︎ the line VignetteBuilder: knitr exists > ☐ any added lines use the same spacing and indents as the existing document > • If agreed with the Maintainer > ☑︎ the contributor is in the author list in the DESCRIPTION file. > ☐ the contributor is in the author list in the vignette’s YAML. > • If this pull request involves converting from separate Author and Maintainer
> lines to the Authors@R vector, please ensure that > ☐ the Authors@R vector includes the maintainer as specified with role='cre'. > ☐ the Maintainer line is completely removed. > ☑︎ HTML document is representative of the PDF in content and
> in general the presentation > ☐ Where the contributor was not able to preserve the content and presentation
> of the PDF is noted as a comment in the pull request > ☐ the R Markdown file is representative of the Sweave document and follows
> best practices, such as replacing links to Bioconductor packages with calls
> to BiocStyle’s Biocpkg() > ☑︎ Only files necessary for the conversion are included in the pull request. > ☑︎ Long lines have been broken up and reformatted, you can achieve this by
> selecting the text in your script and then clicking on Code > Reflow Comment
> in R Studio. > ☑︎ Code blocks are surrounded by a blank line, and there are no blank lines
> before the start or after the end of the code within the code block.
2023-11-14
Federico Marini (14:56:02): > @Federico Marini has joined the channel
2023-12-29
Manvi Yaduvanshi (10:05:01): > @Manvi Yaduvanshi has joined the channel
2024-01-01
Manvi Yaduvanshi (06:40:43): > Hey! I wanted to volunteer for converting vignette. Can I be assigned any vignette so that I can have a hands on it and learn the process?@Sonali Kumari@Andres Wokaty
2024-01-02
Sonali Kumari (23:03:17) (in thread): > Hello@Manvi Yaduvanshi, thanks for inquiring. You can work on this vignette -https://github.com/Bioconductor/annotate/blob/devel/vignettes/useDataPkgs.Rnw. Please share your GitHub user ID with me so that I can update your name on the board. We have a few links pinned in this channel that can be helpful documents for conversion. You can refer to those for converting documents.
Sonali Kumari (23:05:16) (in thread): > After making a PR you can share your update here or in this channel and ask any of the maintainers for review.
2024-01-03
Manvi Yaduvanshi (06:42:40) (in thread): > Thankyou@Sonali Kumari, this is my github profile:https://github.com/manviyadu1shi
2024-01-04
Manvi Yaduvanshi (14:31:14): > While processing the assigned vigenette , when i started knitting , the output came out to be a blank white html page.@Sonali Kumari@Andres Wokaty
Manvi Yaduvanshi (14:31:21): > > processing file: useDataPkgs.Rmd > > output file: useDataPkgs.knit.md > > "C:/Program Files/RStudio/resources/app/bin/quarto/bin/tools/pandoc" +RTS -K512m -RTS useDataPkgs.knit.md --to html4 --from markdown+autolink_bare_uris+tex_math_single_backslash --output useDataPkgs.html --lua-filter "C:\Users\user\AppData\Local\R\win-library\4.2\rmarkdown\rmarkdown\lua\pagebreak.lua" --lua-filter "C:\Users\user\AppData\Local\R\win-library\4.2\rmarkdown\rmarkdown\lua\latex-div.lua" --embed-resources --standalone --variable bs3=TRUE --section-divs --template "C:\Users\user\AppData\Local\R\win-library\4.2\rmarkdown\rmd\h\default.html" --no-highlight --variable highlightjs=1 --variable theme=bootstrap --mathjax --variable "mathjax-url=[https://mathjax.rstudio.com/latest/MathJax.js?config=TeX-AMS-MML_HTMLorMML](https://mathjax.rstudio.com/latest/MathJax.js?config=TeX-AMS-MML_HTMLorMML)" --include-in-header "C:\Users\user\AppData\Local\Temp\RtmpE1G9Yu\rmarkdown-str5e2c44eb5059.html" > [WARNING] This document format requires a nonempty <title> element. > Defaulting to 'useDataPkgs.knit' as the title. > To specify a title, use 'title' in metadata or --metadata title="...". > > Output created: useDataPkgs.html >
Manvi Yaduvanshi (14:31:53) (in thread): > This is the warning I received in terminal. I guess I need a little help out here.
Manvi Yaduvanshi (14:45:57) (in thread): > Update: After a few clicks here and there , I finally got this output : - File (PNG): image.png
{kind=link}
2024-01-05
Andres Wokaty (11:57:55) (in thread): > Hi@Manvi Yaduvanshi! Thanks for working on this vignette. I wonder if the .Rmd has .Rnw that is preventing it from being rendered correctly. I would look above the text “It turns out that the probe id 738…” for the problem. > > If R and R Markdown vignettes (.Rmd) are new to you, I think it might be helpful to look at other vignettes to help you understand the general format. For example, this is R Markdown vignette forhttps://github.com/Bioconductor/BiocStyle/blob/devel/vignettes/AuthoringRmdVignettes.Rmd. > > I also want to recommend that you commit your current work and push your branch to your github repository then I can look at your .Rmd and give you more precise feedback. > > If you’re new togit, you can do this by > > # assuming that you are in annotate/vignettes > # add your .Rmd to the current commit > git add useDataPkgs.Rmd > # make the commit with a message (WIP = "Work in progress") > git commit -m 'WIP create .Rmd' > # push your branch to github.com, where useDataPkgs-Rmd is the name of the branch > git push origin useDataPkgs-Rmd > > When you’ve pushed your branch to GitHub, let me know and I will check out your repository:slightly_smiling_face:
Manvi Yaduvanshi (12:48:29) (in thread): > Sure thing@Andres Wokaty, I’ll commit the changes to the repo for you to review and will check out the example vigenette you’ve linked as well. Thankyou!
2024-01-06
Manvi Yaduvanshi (13:43:02): > Hey!@Andres WokatyI have pushed my changes to my repository but then they were not visible until I created a pull request to the original repo , so my RMD file is now visible as a pull request. If you could have a look and guide me where I’m going wrong, would be a great help. I’m trying to fixing that up myself as well.Update: I guess I’ve figured out the vigenette conversion, I’ll properly make another pull request now to get reviewed.Closing the current one as my doubt is clear now.
Manvi Yaduvanshi (15:35:28): > I’ve made the pull request after resolving the errors.
Andres Wokaty (16:49:11): > Great! I will take a look in the next day or so.
2024-01-09
Sonali Kumari (10:43:30) (in thread): > Hello Reece I wanted to follow up regarding the ongoing conversion. It seems there hasn’t been a pull request from your end yet. Are you still interested in continuing with the conversion, or would you prefer to be removed from the assignee?
Reece (11:16:26) (in thread): > Thank you for the follow up ,let me chime in > A PR soon
2024-01-11
Manvi Yaduvanshi (15:32:04): > @Andres WokatyHey! After the changes you suggested , one of this thing in the date part is throwing error. I tried this code : > > date: `format(Sys.Date(), "%B %d, %Y")` > > but it threw this error: > > Error in yaml::yaml.load(..., eval.expr = TRUE) : > Scanner error: while scanning for the next token at line 3, column 7 found character that cannot start any token at line 3, column 7 > Calls: <Anonymous> ... parse_yaml_front_matter -> yaml_load -> <Anonymous> > Execution halted > > However, if I keep the date blank , eg. > > title: "Using Bioconductor's Annotation Libraries" > author: "Marc Carlson, Jianhua Zhang" > date: > output: > BiocStyle::html_document > > It’s compiling the date as the current date without any error.
Andres Wokaty (15:35:14) (in thread): > This is my fault. It is missing quotes: > > date: "`format(Sys.time(), '%d %B, %Y')`" >
Andres Wokaty (15:35:29) (in thread): > I’m going to update my comment in the PR
Manvi Yaduvanshi (15:43:42) (in thread): > Oh! Okay, gotcha
Manvi Yaduvanshi (15:47:59) (in thread): > Also , i had a few other doubts. Can you please let me know about them? > 1.)Am i supposed to remove the lines about reposTools? - File (PNG): image.png
{kind=link}
Andres Wokaty (15:51:36) (in thread): > Yes, remove the sentence beginning and ending withIf the****reposTools****library ... for both Unix and Windows.
Manvi Yaduvanshi (15:53:45) (in thread): > 2.)You asked me to replace the “>>=” with “}” , and replace the line as > ```
library("annotate") library("hgu95av2.db") library("GO.db")
``` - File (PNG): image.png
{kind=link}
Andres Wokaty (15:54:34) (in thread): > Yes, that’s right.
Manvi Yaduvanshi (15:54:54) (in thread): > It’s throwing some error as : > > Quitting from lines 109-112 [loadLibs] (useDataPkgs.Rmd) > Error in `library()`: > ! there is no package called 'annotate' > Backtrace: > 1. base::library("annotate") > Warning message: > package 'BiocStyle' was built under R version 4.2.1 > > Execution halted >
Manvi Yaduvanshi (15:56:20) (in thread): > I swear that’s the last doubt, I’ve made other changes and they’re hopefully all fine. Really sorry for asking a lot of these :(
Andres Wokaty (15:56:57) (in thread): > oh, soannotateis the package you are working on. What you could do is in an R session, install annotate. The easiest way to do this is viaBiocManager::install("annotate"). That should install annotate and deal with any dependencies.
Manvi Yaduvanshi (15:57:49) (in thread): > Oh! Got it , I’ll just have a run.
Andres Wokaty (15:58:11) (in thread): > No problem! Ask if you have any other questions. Happy to clarify.
2024-01-14
Manvi Yaduvanshi (15:26:00): > @Andres WokatyMade all the suggested changes and opened a pull request, hopefully it’s better now. Can you please have a look?
2024-01-17
Andres Wokaty (10:35:58) (in thread): > I had a brief look, but I think you might have accidentally removed the .Rmd instead of the .Rnw. After you add it back, I will review the PR:slightly_smiling_face:
Andres Wokaty (14:27:58) (in thread): > If you’re having an issue trying to do this, maybe we could set up a time to discuss it or do it together? It’s no problem.
Manvi Yaduvanshi (15:38:12) (in thread): > Oh! my bad, really sorry:face_in_clouds:. I’ll just try adding it again and pull a request asap. If encountered with any problems, I’ll let you know.Thankyou!!
Andres Wokaty (15:38:57) (in thread): > No problem. if you have any issues, we can do a huddle on slack to discuss and do it together.
2024-01-18
Manvi Yaduvanshi (16:04:08) (in thread): > Done with the changes. Hopefully the pull request is done right now:crossed_fingers:
Andres Wokaty (16:05:18) (in thread): > Yes, that looks about right. I will review it tomorrow:slightly_smiling_face:
2024-05-07
Sonali Kumari (11:43:06): - Attachment: Attachment > <!channel> If you were an outreachy applicant and contributed to any of the Bioconductor projects #BugSigDB #sweave2rmd #BSgenomeForge #miaverse, please help us in spreading the word about your amazing work. > > You can share your experience (anonymously) by filling in this short survey https://forms.gle/brqH5M2uTYMKy23x7 > Thank you!
2024-09-29
Mildred Anashie (04:04:43): > @Mildred Anashie has joined the channel
Footnotes
A footnote here.↩︎